【4.1.1】qimme2介绍
一、基本介绍
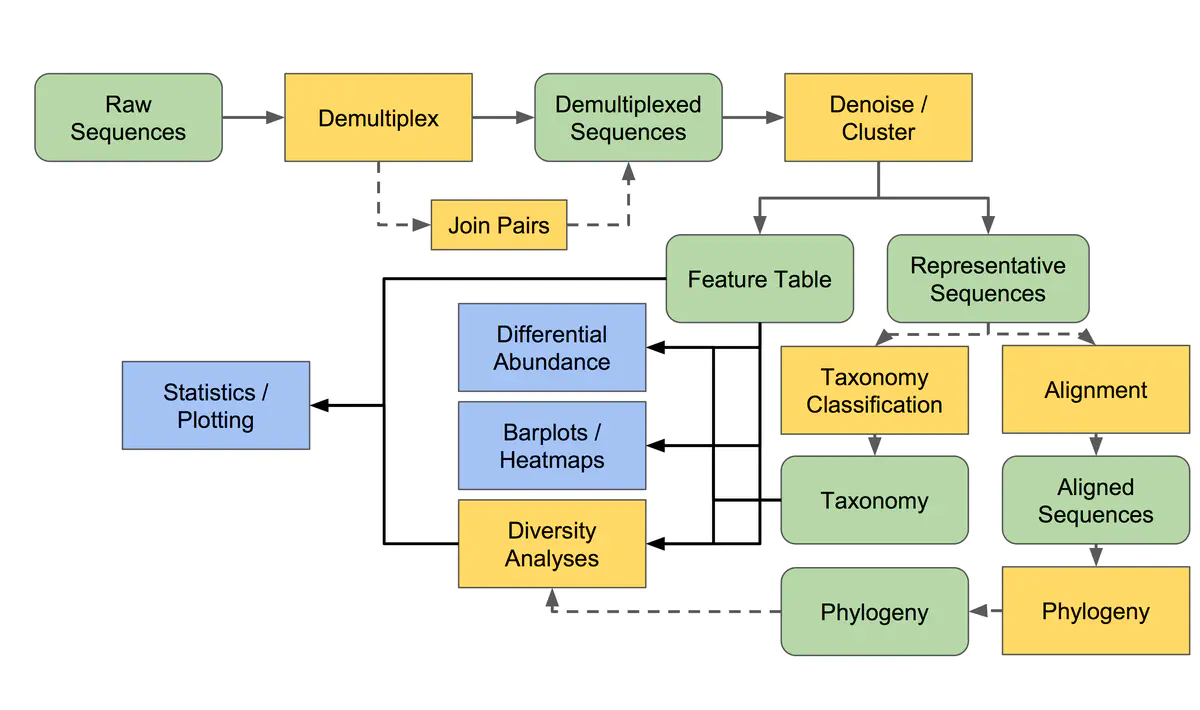
qzv数据的查看
- 可以在 https://view.qiime2.org/ 上查看
QIIME2是微生物组分析流程QIIME的全新版,采用Python3全新编写
- 更易于安装:QIIME2引入了Miniconda软件包管理器,没有管理员权限也可以轻松安装;同时发布了docker镜像,下载即可运行
- 分析流程化:分析流程更加标准化,不让用户盲然下面该做什么;
- 可视化增强:QIIME后发制人,超越引用6964次的mothur流程,就是其可视化方面的优势,现可视化结果更加漂亮,且全新采用交互式图形结果,点选可查看细节,更易于分析
- 方便合作:项目很少一个组可完成,多人多地结果图表方便共享,适合当下科研合作的需求 可扩展:支持自定义功能并加入分析流程;高手可以自己写包,加入QIIME2的流程中
- 分析可重复:全新定义了文件系统,即包括分析数据、也包括分析过程和结果,每一步的结果,均可追溯全部分析过程,方便检查和重复
三、qiime2功能介绍
Usage: qiime [OPTIONS] COMMAND [ARGS]...
Commands:
info Display information about current deployment.
tools Tools for working with QIIME 2 files.
dev Utilities for developers and advanced users.
alignment Plugin for generating and manipulating alignments.
composition Plugin for compositional data analysis.
cutadapt Plugin for removing adapter sequences, primers, and other unwanted sequence from sequence data.
dada2 Plugin for sequence quality control with DADA2.
deblur Plugin for sequence quality control with Deblur.
demux Plugin for demultiplexing & viewing sequence quality.
diversity Plugin for exploring community diversity.
emperor Plugin for ordination plotting with Emperor.
feature-classifier Plugin for taxonomic classification.
feature-table Plugin for working with sample by feature tables.
fragment-insertion Plugin for extending phylogenies.
gneiss Plugin for building compositional models.
longitudinal Plugin for paired sample and time series analyses.
metadata Plugin for working with Metadata.
phylogeny Plugin for generating and manipulating phylogenies.
quality-control Plugin for quality control of feature and sequence data.
quality-filter Plugin for PHRED-based filtering and trimming.
sample-classifier Plugin for machine learning prediction of sample metadata.
taxa Plugin for working with feature taxonomy annotations.
vsearch Plugin for clustering and dereplicating with vsearch.
3.1 数据导入
Usage: qiime tools import [OPTIONS]
Options:
--type TEXT 每步分析都会产生qza文件,都会有相应的语义类型,避免用户不合理的分析过程。
--input-path PATH 输入文件路径,文件格式为sample-id,absolute-filepath,direction
--output-path ARTIFACT 输出文件:.qza格式的文件名
--input-format TEXT 输入数据格式:双端测序数据可设置为PairedEndFastqManifestPhred33V2
--show-importable-types 查看所有的语义类型
--show-importable-formats 查看所有输入数据格式
--help 查看说明
示例
# 导入原始序列
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \ #成对的带质量序列,要求序列ID与样品编号存在对应关系
--input-path sample_list.txt \
--input-format PairedEndFastqManifestPhred33 \
--output-path demux-paired-end.qza
# 导入.biom格式的特征表:转换成qza格式
qiime tools import \
--type 'FeatureTable[Frequency]' \ # FeatureTable[Frequency]: 频率,即Feature表(OTU表),为每个样品中对应OTU出现频率的表格
--input-path feature_table.biom \
--input-format BIOMV210Format \
--output-path feature_table.qza
3.2 生成OTU表
Usage: qiime dada2 [OPTIONS] COMMAND [ARGS]...
主要作用是去除低质量序列、嵌合体;再生成OTU表,现在叫Feature表,因为不再使用聚类方法,相当于QIIME时代100%相似度的OTU表。
Options:
--version Show the version and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
denoise-paired Denoise and dereplicate paired-end sequences
denoise-pyro Denoise and dereplicate single-end pyrosequences
denoise-single Denoise and dereplicate single-end sequences
示例
qiime dada2 denoise-paired \
--i-demultiplexed-seqs demux-paired-end.qza \
--o-table feature_table.qza \
--o-representative-sequences rep-seqs-dada2.qza \ #输出代表性序列文件
--o-denoising-stats stats-dada2.qza \ #过程统计
--p-trim-left-f 0 \ #表示从forward左端的第0位置开始
--p-trim-left-r 0 \ #表示从reverse左端的第0位置开始
--p-trunc-len-f 250 \ #表示forward总共保留的长度为250
--p-trunc-len-r 250 #表示reverse总共保留的长度为250
3.3 操作特征表
Usage: qiime feature-table [OPTIONS] COMMAND [ARGS]...
Description: This is a QIIME 2 plugin supporting operations on sample by
feature tables, such as filtering, merging, and transforming tables.
Commands:
core-features Identify core features in table
filter-features Filter features from table
filter-samples Filter samples from table
filter-seqs Filter features from sequences
group Group samples or features by a metadata column
heatmap Generate a heatmap representation of a feature table
merge Combine multiple tables
merge-seqs Combine collections of feature sequences
merge-taxa Combine collections of feature taxonomies
presence-absence Convert to presence/absence
rarefy Rarefy table
relative-frequency Convert to relative frequencies
subsample Subsample table
summarize Summarize table
tabulate-seqs View sequence associated with each feature
transpose Transpose a feature table.
特征表统计
qiime feature-table summarize \
--i-table feature_table.qza \
--o-visualization feature_table.qzv
# QIIME2生成的图表结果文件类型,以.qzv为扩展名,末尾的v代表visual
过滤特征表:过滤样本
qiime feature-table filter-samples \
--i-table feature_table.qza \
--m-metadata-file sample-to-keep.tsv \
--o-filtered-table feature_table_filtered.qza
3.4 多样性分析
Usage: qiime diversity [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin supports metrics for calculating and
exploring community alpha and beta diversity through statistics and
visualizations in the context of sample metadata.
Commands:
adonis adonis PERMANOVA test for beta group significance
alpha Alpha diversity
alpha-correlation Alpha diversity correlation
alpha-group-significance Alpha diversity comparisons
alpha-phylogenetic Alpha diversity (phylogenetic)
alpha-rarefaction Alpha rarefaction curves
beta Beta diversity
beta-correlation Beta diversity correlation
beta-group-significance Beta diversity group significance
beta-phylogenetic Beta diversity (phylogenetic)
beta-rarefaction Beta diversity rarefaction
bioenv bioenv
core-metrics Core diversity metrics (non-phylogenetic)
core-metrics-phylogenetic Core diversity metrics (phylogenetic and non-
phylogenetic)
filter-distance-matrix Filter samples from a distance matrix.
mantel Apply the Mantel test to two distance matrices
pcoa Principal Coordinate Analysis
pcoa-biplot Principal Coordinate Analysis Biplot
procrustes-analysis Procrustes Analysis
核心多样性(无系统发育树)
qiime diversity core-metrics \
--i-table feature_table.qza \
--p-sampling-depth 138000 \
--m-metadata-file sample_metadata.tsv \
--output-dir /data/hushy/XJ/data_BFV/qiime2/diversity #自动创建目录
# alpha多样性observed_otus(OTU数量)指数 observed_otus_vector.qza
# alpha多样性香农熵(考虑物种和丰度)指数 shannon_vector.qza
# alpha多样性evenness(均匀度,考虑物种和丰度)指数 evenness_vector.qza
# beta多样性bray_curtis距离矩阵 bray_curtis_distance_matrix.qza
# beta多样性可视化结果bray_curtis_emperor.qzv,图中Axis1 表示尽可能最大解释数据变化的主坐标成分,Axis2 为解释余下的变化度中占比例最大的主坐标成分,Axis3 等依次类推。
#Richness, Chao1,Shannon三个指数是常用的评估丰富度的指标,数值越高表明样品包含的物种丰富度就越高。
#Jaccard距离(群落差异的定性度量,即只考虑种类,不考虑丰度)
#Bray-Curtis距离(群落差异的定量度量)
#非加权UniFrac距离(包含特征之间的系统发育关系的群落差异定性度量)
#加权UniFrac距离(包含特征之间的系统发育关系的群落差异定量度量)
alpha多样性可视化及组间显著性分析
qiime diversity alpha-group-significance \
--i-alpha-diversity shannon_vector.qza \
--m-metadata-file sample_metadata.tsv \
--o-visualization shannon_vector.qzv
药企,独角兽,苏州。团队长期招人,感兴趣的都可以发邮件聊聊:tiehan@sina.cn
![]() 个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
