【7.4.3】 挖掘天然产物生物合成基因簇的宏基因组--BiG-MEx
软件地址:https://github.com/pereiramemo/BiG-MEx R包:https : //github.com/pereiramemo/bgcpred
-
微生物通过生物合成基因簇 (BGC) 的表达产生种类繁多的天然产物:物理成簇的基因编码特定代谢途径的酶。这些天然产品涵盖广泛的化学类别(例如,氨基糖苷类、羊毛硫抗生素、非核糖体肽、低聚糖、聚酮化合物、萜烯),对工业和医疗应用1具有很高的价值。
-
在这里,我们通过开发超快生物合成基因簇 MEtagenomic eXploration 工具箱 (BiG-MEx) 来应对这一挑战。BiG-MEx 快速识别广泛的 BGC 蛋白结构域,评估它们的多样性和新颖性,并预测宏基因组数据中天然产物 BGC 类别的丰度概况
-
我们展示了与标准 BGC 挖掘方法相比 BiG-MEx 的优势,并使用它来探索 TARA 海洋和人类微生物组项目数据集4中样本的 BGC 域和类别组成。在这些分析中,我们证明了 BiG-MEx 在宏基因组数据中研究 BGC 的分布、多样性和生态作用的适用性,并指导具有临床应用的天然产物的探索。
一、前言
宏基因组学为在来自各种环境的不同微生物组合中挖掘天然产物 BGC 提供了独特的机会5 – 7. 然而,鉴于自然界中发现的微生物群落的复杂性,以及当前测序技术的局限性,通常只能将一小部分短读序列数据组装到足够长的重叠群中,以识别 BGC 类别。然而,BGC 的单个蛋白质结构域的注释要直接得多,因为它们具有与合并的双端读取相当的长度。已知有几个蛋白质结构域在 BGC 编码的酶中发挥重要作用。在某些类型的 BGC 类中通常可以找到特定域或其组合。因此,这些用于自动识别基因组序列8 – 10 中的 BGC 类别并研究特定 BGC 类别在环境中的分布和多样性6 , 7 , 11 – 13。尽管有各种具有实际应用的 BGC 挖掘工具14,但只有自然产品领域寻求者 (NaPDoS) 11和自然产品多样性环境调查员 (eSNaPD 15) 致力于 BGC 域的研究。这两种工具都专注于非核糖体肽和聚酮化合物合酶(分别为 NRPS 和 PKS),并将组装或扩增子数据作为输入。目前,还没有能够有效利用原始宏基因组数据来研究环境中天然产物 BGC 类别和域的组成和多样性的技术。
利用 BGC 域可以在未组装的宏基因组数据中轻松注释的事实,并用于识别不同的天然产物 BGC 类别,我们开发了 BiG-MEx。该工具在短读序列数据中生成超快 BGC 域注释,并应用机器学习方法来预测基于 BGC 类覆盖的丰度(为简单起见,我们将这些称为 BGC 类丰度概况)。此外,识别的域序列用于执行基于域的多样性分析。这使得 BiG-MEx 既能深入利用宏基因组数据,又能适应其不断增加的数据量。BiG-MEx 由三个交互模块组成,如下所述并如图 1 所示:
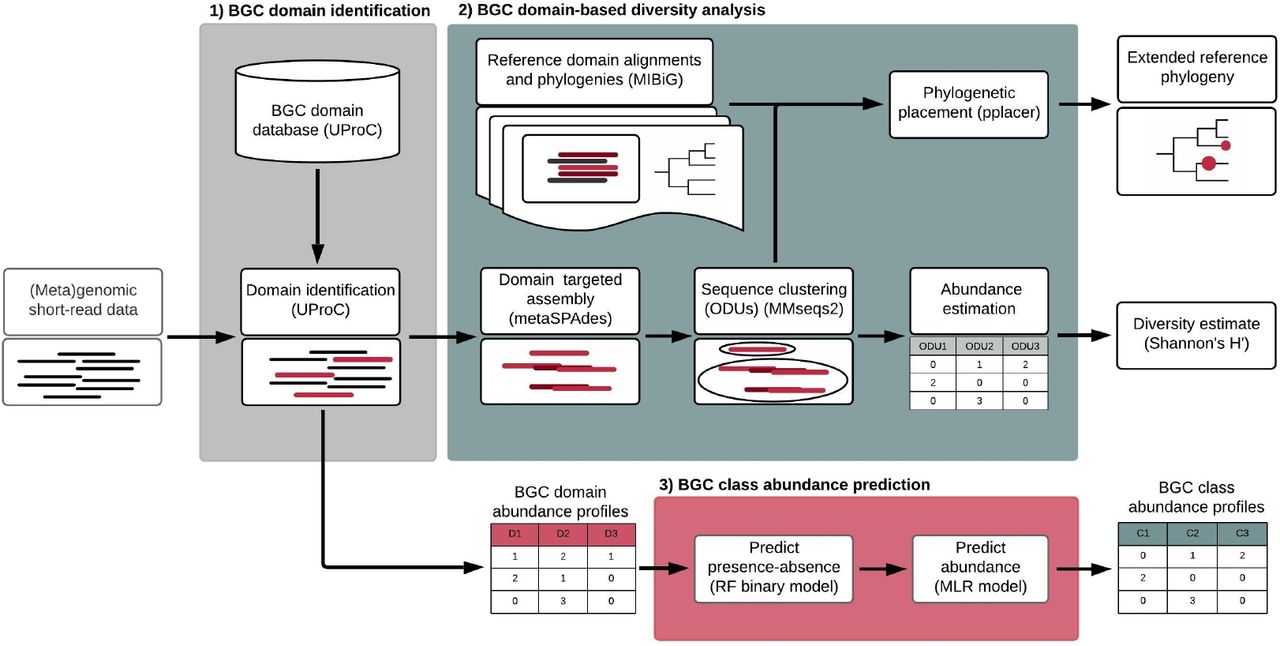
图1 Big-MEx 分析工作流程。
- BGC域识别模块。为了用 UProC 注释 BGC 域,我们创建了一个包含 150 个域的 UProC 数据库,这些域来自 44 个不同的 BGC 类。该数据库是基于 antiSMASH 隐马尔可夫模型 (HMM) 配置文件10的氨基酸序列生成的。使用 UProC 输出,该模块生成基于计数的 BGC 域丰度概况;2)基于BGC域的多样性分析模块。使用先前确定的域,该模块使用 metaSPAdes 45执行目标组装重构域序列。组装的域序列被聚类为操作域单元,并使用ODU的数量和每个ODU内域序列的覆盖范围(用于近似ODU的丰度)来计算ODU alpha多样性。环境重建域序列被放置在具有 pplacer 48(最大似然标准)的参考系统发育树上。在这个模块中,我们包括 48 个域的预先计算的系统发育,这些域基于生物合成基因簇的最小信息 (MIBiG) 51数据库中包含的序列数据,使我们能够识别查询序列与来自以下途径的域的关系已知功能;3)BGC类丰度预测模块。域丰度概况用于使用特定于类的机器学习模型预测基于 BGC 类覆盖的丰度概况。这些模型由两步过程组成:首先,使用随机森林 (RF) 分类器预测 BGC 类的存在/不存在;其次,仅当该类别先前被预测为存在时,才使用多元线性回归 (MLR) 预测丰度。
模块:
-
BGC域名识别模块。我们使用超快蛋白质域分类 UProC 16工具来识别短读序列数据中的 BGC 蛋白质域。为此,我们创建了一个 UProC 数据库,其中包含 150 个 BGC 域,涵盖 44 个 BGC 类。
-
基于 BGC 域的多样性分析。该模块执行以域为目标的组装,将组装的域序列聚类以创建操作域单元 (ODU) 17并计算 ODU alpha 多样性。此外,组装的域序列被放置在参考系统发育树上。该模块包括 48 个 BGC 域的预计算系统发育。这些是基于来自实验表征的生物合成基因簇的域序列选择的,具有足够的序列信息进行系统发育分析。
-
BGC类丰度预测模块。我们创建了机器学习模型,可以根据域注释预测 BGC 类的丰富程度。这些模型是特定于类的,由一个随机森林 (RF) 分类器来预测 BGC 类的存在/不存在,以及一个多元线性回归 (MLR) 来预测其丰度。这些模型可以定制,以分别针对来自不同环境和分类群的宏基因组和基因组数据。
二、结果
略
三、方法
3.1. 数据采集、预处理和标注
- 我们从欧洲核苷酸档案(ENA:PRJEB1787,过滤器大小:0.22-1.6 和 0.22-3)中检索了 TARA Oceans 数据集的 139 个原核宏基因组。
- 为了预处理宏基因组短读数据,我们使用 BBMap 35.00 套件 ( https://sourceforge.net/projects/bbmap / ),最大汉明距离为 1 (hdist=1)。
- 然后我们使用 VSEARCH 2.3.4 31合并配对末端读数,质量在 Q20 修剪所有读数并使用 BBDuk 过滤掉短于 45bp 的序列,并使用 VSEARCH 去复制得到的质量控制序列。
- 我们通过首先使用 FragGeneScan-plus 预测预处理数据中的开放阅读框 (ORF) ,然后在预测的 ORF 的氨基酸序列上运行 BiG-MEx 来注释 BGC 域。
我们从人类微生物组计划 (HMP) 的数据分析和协调中心 (DACC) 下载了 491 个人类微生物组宏基因组 ( https://www.hmpdacc.org/hmp/HMASM/ )。我们的数据集包括龈上斑块 (118)、舌背 (128)、颊粘膜 (107) 和粪便 (138) 身体部位的宏基因组。这些宏基因组已经按照 The Human Microbiome Project Consortium 2012 中的描述进行了预处理. 我们执行的额外预处理任务包括将宏基因组读数与 VSEARCH 合并、在 Q20 质量修剪所有读数以及使用 BBduk 过滤掉短于 45 bp 的序列。为了注释 BGC 域,我们用 FragGeneScan-plus 预测了 ORF,并在 ORF 的氨基酸序列上运行了 BiG-MEx BGC 域识别模块(补充表 7)
3.2 对 TARA Oceans 和 HMP 数据集进行的探索性分析
使用 TARA Oceans 和 HMP 宏基因组的域丰度图谱,通过 BiG-MEx BGC 类丰度预测模块预测 BGC 类丰度图谱。用于生成 TARA 海洋预测的模型以及口腔和粪便 HMP 宏基因组分别使用 Marine-RM、人类-口腔和人类-粪便模拟宏基因组数据集进行训练。对于每个数据集,我们执行如下主坐标分析 (PCoA):
- 我们对域和类丰度矩阵应用了总和缩放标准化;
- 我们使用标准化矩阵来计算域和类 Bray-Curtis 相异矩阵;
- 我们使用使用 vegan R 包的函数 capscale 对相异矩阵执行 PCoA 。
我们应用了方差排列多变量分析 (PERMANOVA) 来量化强度并根据 BGC 类组成测试水层和身体部位之间的差异。对于这些分析,我们从 TARA Oceans 和 HMP 数据集(分别为 63 和 216 个宏基因组;见下文)中选择了平衡的宏基因组子集。我们对如上所述为 TARA 海洋和 HMP 宏基因组子集计算的 Bray-Curtis 相异矩阵执行了 PERMANOVA,以同时测试所有组之间的差异。随后,我们独立测试了每对组,应用 Bonferroni 校正进行多重比较。为了执行 PERMANOVA,我们使用了 vegan R 包的 adonis 函数,排列参数设置为 999。
为了比较表面 (SRF)、深叶绿素最大值 (CDM) 和中层 (MES) 水层之间 NRPS 腺苷酸化(AMP 结合)和凝聚域的域 ODU 多样性,我们使用了 63 个 TARA 海洋宏基因组的子集,代表21个采样站的三个水层。我们使用在基于 BiG-MEx 域的多样性模块中实现的例程计算了这些宏基因组中的 ODU Shannon 多样性。此外,我们使用相同的 BiG-MEx 模块来检查代表采样站 TARA_085 的三个水层的宏基因组中凝结域的多样性。为了执行 ODU 分类法注释,我们使用了基于 UniRef100 的 MMseqs2 分类法分配函数序列 (release-2018_08),e 值和灵敏度参数分别设置为 0.75 和 0.01。为了比较身体部位之间的 AMP 结合和凝聚 ODU 多样性,我们应用了如上所述的类似方法。我们选择了 216 个宏基因组的子集,其中 54 个来自龈上斑块、舌背、颊粘膜和粪便体部位。该子集仅包括从四个身体部位采样的个体中获得的宏基因组。我们应用了基于 BiG-MEx 域的多样性模块来计算 ODU Shannon 多样性估计。
Wilcoxon 秩和检验(两侧)用于评估来自不同组(即水层或身体部位)的宏基因组之间差异的显着性,使用来自 R 包 stats 38的 wilcox.test 函数进行。
3.3 数据模拟、预处理和标注
3.3.1 模拟宏基因组数据集的构建
….
我们使用 MetaSim v0.9.5 41模拟了宏基因组。MetaSim 被设置为生成长度为 101bp 的双端读数,并且替换率沿着每个读数从 1×10 -4到 9.9×10 -2 不断增加。利用这些数据,我们旨在模拟 Illumina HiSeq 2000 平台生成的短读序列。
3.3.2 模拟宏基因组的注释
为了估计模拟宏基因组中参考 BGC 类的丰度,
- 我们使用 antiSMASH 3.0 注释其参考基因组序列中的 BGC 类,
- 使用 BWA-MEM 0.7.12 将配对末端读数映射到已识别的 BGC 序列,并过滤掉读数质量分数低于 10 的对齐。
- 接下来,我们使用 Picard 工具 v1.133 ( http://broadinstitute.github.io/picard )删除重复reads,
- 并使用 BEDtools v2.23 计算平均覆盖率. 假设覆盖率估计准确反映了基于 BGC 类覆盖率的丰度,因为它们是使用完整的 BGC 序列计算的,从用于模拟宏基因组的基因组序列中获得。
- 此外,我们将模拟宏基因组的配对末端读数与 VSEARCH 2.3.4 合并,使用 FragGeneScan-plus 预测 ORF,并使用 BiG-MEx 域识别模块对 ORF 氨基酸序列中的 BGC 域进行注释。
可以在 https://github.com/pereiramemo/BiG-MEx/wiki/Data-simulation#7-bgc-domain-annotation 找到注释合成宏基因组的工作流程
3.4. 绩效评估
3.4.1 BGC域识别模块
我们将 UProC (即 uproc-prot)的运行时间(挂钟)与使用来自 HMMER3 包的 hmmsearch 的典型搜索进行比较,以识别包含在 BiG-MEx 中的 9 个原核生物宏基因组中的 150 个 BGC 域。 TARA Oceans 数据集(补充表 8)。为了运行 hmmsearch,我们使用了 antiSMASH 的域 HMM 配置文件。我们在四轮独立的轮次中使用这两种工具对九个宏基因组进行了注释,每轮使用不同的线程数(即,4、8、16 和 32 个线程)。uproc-prot 和 hmmsearch 的所有参数都设置为默认值。注释是在配备 Intel(R) Xeon(R) CPU E7-4820 v4 2.00GHz 处理器的工作站上进行的。
3.4.2 基于BGC域的多样性分析模块
我们使用 NRPS 腺苷酸化(AMP 结合)和缩合以及 PKS 酮合酶和酰基转移酶域(分别为 PKS_KS 和 PKS_AT)评估了 BiG-MEx 操作域单元 (ODU) 多样性估计方法。在这个分析中,我们使用 BGC 基于域的多样性分析模块来计算 Marine-TM 数据集中的 ODU 多样性,并将这些估计与使用完整域序列计算的 ODU 多样性进行比较。为了获得后者的 ODU 多样性,我们应用了在 BiG-MEx 中实现的工作流程,除了我们从用于模拟 Marine-TM 宏基因组的完整基因组序列中提取这些序列,而不是组装域序列。我们使用 antiSMASH HMM 配置文件使用 hmmsearch 注释了完整基因组序列中的四个域。
3.4.3 BGC类丰度预测
我们使用用 Marine-RM 宏基因组数据集训练的 BGC 类模型来预测 Marine-TM 宏基因组数据集中的 BGC 类丰度。我们应用 3.2 节中描述的方法来计算基于完整基因组序列(即参考丰度)的 Marine-TM 宏基因组中的 BGC 类丰度。为了使用机器学习模型预测 BGC 类丰度,我们使用 BiG-MEx 域识别模块对 Marine-TM 宏基因组进行了注释,并将域丰度配置文件用作 BiG-MEx BGC 类丰度预测模块的输入。评估包括计算预测和参考 BGC 类丰度之间的 Pearson 相关性和中值无符号平方误差 (MUE)。MUE 计算为 | Â - A |/ A,其中Â和A分别是预测丰度和参考丰度。为了对机器学习模型进行基准测试,我们将 BGC 类丰度预测与基于 Marine-TM 数据集的 50 个宏基因组的组装(组装方法)的丰度估计进行了比较。组装方法包括使用 MEGAHIT(默认参数)组装宏基因组,运行 BiG-MEx 域识别模块以选择具有潜在 BGC 序列的重叠群,使用 antiSMASH 3.0 注释所选重叠群,并按照与以下相同的方法估计 BGC 类丰度在第 3.2 节(补充表 9)中描述。我们计算了无符号误差,以及通过组装方法获得并由 BiG-MEx 预测的 BGC 类丰度估计值的 Pearson 相关系数,相对于参考 BGC 类丰度。 https://rawgit.com/pereiramemo/BiG-MEx/master/machine_leaRning/bgcpred_workflow.html
3.4.4 浅层宏基因组中 BGC 类丰度预测的评估
我们选择了 30 个合并的预处理 TARA Oceans 宏基因组,并使用 seqtk v1.0 工具( https://github.com/lh3 /seqtk )。然后,我们使用 BiG-MEx(如第 1 节和第 2 节所述)注释了 BGC 域并预测了该数据中的 BGC 类丰度,并比较了两组宏基因组之间的 BGC 类丰度预测。
3.5. Big-MEx 实施
3.5.1 BGC域识别模块
BiG-MEx BGC 域识别模块使用 UProC 1.2.0 16软件对使用 BGC 域引用的短读序列进行分类。为了为此目的训练 UProC,我们手动整理了与 150 个 antiSMASH 隐藏马尔可夫模型配置文件 (HMM) 10匹配的所有氨基酸序列. 在这项任务中,我们删除了短于 25 个氨基酸的序列,并检查了不同 HMM 谱的序列之间是否存在重叠。此外,我们将多域蛋白质分为多个家族。在训练过程中,我们包含了一组阴性对照配置文件来评估假阳性命中率。即,我们使用 t2fas、fabH、bt1fas、ft1fas 配置文件作为 PKS_KS、t2ks、t2ks2、t2clf、Chal_sti_synt_N、Chal_sti_synt_C、hglD 和 hglE 配置文件的阴性对照。一旦我们整理了氨基酸序列数据,我们就应用了来自 NCBI Blast+ 2.2 Suite 44的 SEG(mentation) 低复杂性过滤器并创建了 UProC 数据库。这个 UProC 数据库可以从 https://github.com/pereiramemo/BiG-MEx 下载. 基于识别出的包含 BGC 域序列的读取,该模块计算 BGC 域的基于计数的丰度概况。
3.5.2 基于BGC域的多样性分析模块
该模块执行两种不同的分析:操作域单元 (ODU) 多样性估计和域序列的系统发育位置。估计 ODU 多样性的管道,独立分析每个域,包括以下步骤:
- 短读序列,其中正在研究的域被识别,被招募来执行具有默认参数的目标组装 metaSPAdes 3.11 45;
- 使用 FragGeneScan-Plus 预测结果重叠群中的开放阅读框 (ORF);
- 从HMMER v3中用hmmsearch在ORF氨基酸序列内鉴定域序列并提取;
- 域氨基酸序列使用 MMseqs2 46聚类成 ODU使用级联聚类选项并将灵敏度参数设置为 7.5;
- 使用BWA-MEM 0.7.12将带注释的未组装reads映射到域核苷酸序列,使用BEDtools v2.23计算平均深度覆盖率;
- 基于此信息,计算基于覆盖范围的 ODU 丰度并用于估计 ODU alpha 香农多样性。
为了比较不同测序深度的样本之间的 ODU 多样性估计,我们提供了一个选项来估计稀疏子样本的多样性。
- 为了执行域序列的系统发育放置,我们应用了类似于 NaPDoS 的方法。然而,我们将系统发育定位分析扩展到 48 个域,并包括更全面的参考树,这对于分析大型宏基因组样本至关重要。
- 详细地说,系统发育放置包括使用 MAFFT (使用 –add 选项)将目标域序列与其相应的参考多序列比对 (MSA) 进行比对。
- 随后,扩展的 MSA 及其参考树被用作运行 pplacer 的输入(带参数:–keep-at-most 10 和–discard-nonoverlapped;所有其他参数设置为默认值)。pplacer 使用最大似然标准执行系统发育放置,并以 Newick 和 jplace 格式输出扩展树,以及包含有关每个序列放置的统计信息和信息的表格(即,似然性、后验概率、放置位置之间的预期距离( EDPL)、垂饰长度和边数)。
- 为了可视化系统发育位置,使用 ggtree R 包生成树图,其中放置序列的覆盖范围被映射到它们的树尖上,并用于缩放气泡表示。除了系统发育位置,我们在这个模块中包含了一个选项,用于针对参考域序列对组装的域序列执行 BLASTP 搜索。
为了构建参考系统发育,
- 我们首先从 MIBiG 数据库中下载了所有 BGC 氨基酸序列.
- 我们使用来自 antiSMASH 的 BGC 域 HMM 配置文件通过 hmmsearch 识别域序列。
- 随后,我们使用 MMseqs2 提取和聚类这些序列,为每个域创建一个非冗余的氨基酸序列数据集。
- 如果 MIBiG 数据库中鉴定的参考序列数量大于 500,我们在氨基酸水平上使用 0.7 同一性的聚类阈值;否则,阈值设置为 0.9;MMseqs2 的所有其他参数都按照之前的规定进行设置。丢弃少于 20 个代表性序列的所有域。这导致考虑用于系统发育重建的 48 个域的子集。对于每组域代表序列,我们使用 E-INS-I 算法生成带有 MAFFT 的 MSA,使用 OD-seq 去除序列异常值并用 RAxML 构建了系统发育树。
为了选择用于系统发育重建的蛋白质进化模型,我们使用了在 RAxML 中实现的具有最大似然标准的自动模型选择。我们使用速率异质性的 GAMMA 模型并使用快速爬山算法搜索树空间,从最大简约树开始。为了重现性,我们指定了一个随机种子数(即 -p 12345)。最后,我们使用 RAxML 来根树并计算类似 SH 的支持分数. 在补充文件 2 中,我们为每个域系统发育提供了使用的序列数量和氨基酸替代模型、平均值、标准偏差、序列之间的最大和最小共表距离、Faith 的系统发育多样性 及其相应 BGC 类的名称。。
3.5.3 BGC类丰度预测模块
BiG-MEx 使用机器学习模型根据未组装宏基因组中注释域的计数来预测 BGC 类的丰度。每个模型都是特定于类别的,并使用 BGC 类别的丰度及其相应的蛋白质域分别作为响应和预测变量进行训练。我们使用 antiSMASH 中定义的分类规则来注释 BGC 类别,以确定在每个模型中用作预测变量的蛋白质域。为了模拟给定 BGC 类的丰度,我们实施了一个两步零膨胀过程。首先,使用随机森林(RF)二元分类器预测目标 BGC 类的存在与否56. 其次,应用多元线性回归 (MLR) 来预测类别丰度,但前提是该类别先前被预测为存在。在零值的数量低于 10 或不存在的情况下,我们直接应用 MLR。我们使用模拟宏基因组数据(即 Marine-RM、Human-Oral 和 Human-Stool 数据集)训练模型。模型预测基于覆盖的丰度,因为这是训练过程中使用的响应变量。RF 二进制分类模型是使用 randomForest R 包57的 randomForest 函数创建的,参数 ntree 设置为 1000(生长的树的数量),nodesize 设置为 10(终端节点的最小大小),并且 mtry 设置为 1(在每次拆分时随机采样作为候选的变量数)。对于 MLR,我们使用带有默认参数的 stats R 包 ( https://www.R-project.org/ )的 lm 函数。
参考资料
- Mining metagenomes for natural product biosynthetic gene clusters: unlocking new potential with ultrafast techniques。 https://www.biorxiv.org/content/10.1101/2021.01.20.427441v1.full
