【5.2.1.3】基因本体论与GO注释
一、本体论和基因本体论
不同的生物学家研究不同类但相似的基因的时候,需要定义一套共同的语言:
- 如果是同源基因,你希望它的名字,同义词还有一些特征都有明确的定义,不管你研究那个物种,你一看就能明白。
- 家族信息也需要定义,这就是relationship的一个定义。
这样定义出来,即是一套词库,就是一个vocabulary,他不是一个普通的vocabulary,而是一个controled vocabulary,就是会说一旦定义好了这个词库对于这些基因的注释里,今后就不能随心所欲的用自己想要的词,而是要从这个词库里来选择已经订好的固定的词。同时它也是一个common vocabulary,就是说这几个组织研究不同的物种同意来定义一些生物学的共同的概念,common controlled vocabulary.它是hierarchical,就是有结构的,这个结构是通过定义的家族从属关系,比如说complex subunit的从属关系就是通过这些来实现的,这样的一个东西,如果要做得非常非常规范的时候,他就可以被称为一个ontology,中文称为本地论。
本体论,A specification of conceptualization,就是说针对你所研究的这个领域,你要定义一系列的概念,这些概念有一个共同的词库。这个词库定义了两件事情,一个是领域中所有的entity的名称,类型和特征,同时你要定义概念之间的关系。Ontology这个概念最初并非计算机领域的概念,虽然我们提到它在计算机尤其是人工智能里应用地比较个广,但是最早Ontology是一个哲学的概念。她定义的是研究存在和现实,the nature of being ,becoming,existence or reality,它的主要类型和他们之间的关系计算机是借助了这样一个哲学的概念使它变成一个可操作的概念。
在生物中的应用,它能辅助不同生物学家和不同人之间的交流,另一个是能够注释基因组的时候有一套共同的语言。对于生物信息学十分重要的是使我们能够做计算,因为数据存在电脑里,如果都是free-text,也就是完全的自由文本文件的话,很多统计分析分析是没有办法做的,但如果你的数据定义底层有一套严格的ontology的话,那就可以用各种统计方法做很多计算,所以,ontology enables compution.第三,ontology的结构是非常重要的,它可以让我们在电脑自动地找到一些超越单个基因之上的大的模式。如果有了这样的ontology定义之后,任何一组基因都可以看有没有一些大的功能类别或者是pathway。它等于是给了你一个所谓的bird eyeview.
在生物学里,有若干用的比较多的ontology,包括gene ontology,anatomical entity ontology,disease ontology, sequence ontology,system biology.这里用的最多。大家最应该知道的就是Gene ontology.
Gene Ontology在最开始做果蝇基因组,酵母基因组测序,小鼠基因组测序这三个比较大的项目时,Genome Centre决定建立起来的,它在1998年时开始启动,就是在不同物种之间有一定同源性的这些基因和基因产物,包括蛋白,也包括一些非编码RNA,描述他们的一套structured,hierarched common controlled vocabulary.到今天参与Gene Ontology的机构从最初的三家发展到了将近20家,可以看到已经涵括了很多个物种。Gene Ontology为了描述基因和基因产物,它定义了三个大的类别,因为大家也知道任何一个基因都可以从分子功能,所参与的生物过程,处的亚细胞的定位几个方面来分别的定义。分子功能定义的是基因产物它所行使的任务,比如carbohydrate binding和ATPase activity. Biological Process是更广一点所参与的生物过程,比如说mitosis,purine metabolism.Cellular component 就是它的亚细胞定位,它的聚合体等等。
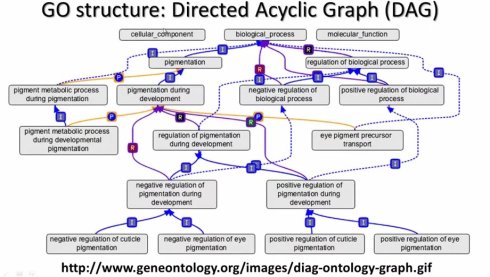
GO的hierarchical structure如果画出来就是这个样子,最根部是三颗树,就是这三大类,是一个有向无环图。它主要有几种存储模式,一种就是OBO文件格式,OBO是把里面的每一个概念定义为一个Term,每一个Term有一系列的特征,包括:unique ID,名字,namespace,definition,synonym同义词,is a从属关系。另一个格式就是XML格式,XML格式有点像HTML格式,把每个概念定义为一个TERM,每个TERM也有一个accession number,有它的名字,同义词还要定义,另外iaruguo这个基因在别的数据存在,也有链接存储在里面,所XML文件在电脑里,以go:accession开始,o:accesion结束,
Gene Ontology可分为:
- 分子功能(Molecular Function)
- 生物过程(biological process)
- 细胞组成(cellular component)
蛋白质或者基因可以通过ID对应或者序列注释的方法找到与之对应的GO号,而GO号可对于到Term,即功能类别或者细胞定位。
功能富集分析: 功能富集需要有一个参考数据集,通过该项分析可以找出在统计上显著富集的GO Term。该功能或者定位有可能与研究的目前有关。 GO功能分类是在某一功能层次上统计蛋白或者基因的数目或组成,往往是在GO的第二层次。此外也有研究都挑选一些Term,而后统计直接对应到该Term的基因或蛋白数。结果一般以柱状图或者饼图表示。
- GO分析 根据挑选出的差异基因,计算这些差异基因同GO 分类中某(几)个特定的分支的超几何分布关系,GO 分析会对每个有差异基因存在的GO 返回一个p-value,小的p 值表示差异基因在该GO 中出现了富集。 GO 分析对实验结果有提示的作用,通过差异基因的GO 分析,可以找到富集差异基因的GO分类条目,寻找不同样品的差异基因可能和哪些基因功能的改变有关。
- Pathway分析 根据挑选出的差异基因,计算这些差异基因同Pathway 的超几何分布关系,Pathway 分析会对每个有差异基因存在的pathway 返回一个p-value,小的p 值表示差异基因在该pathway 中出现了富集。 Pathway 分析对实验结果有提示的作用,通过差异基因的Pathway 分析,可以找到富集差异基因的Pathway 条目,寻找不同样品的差异基因可能和哪些细胞通路的改变有关。与GO 分析不同,pathway 分析的结果更显得间接,这是因为,pathway 是蛋白质之间的相互作用,pathway 的变化可以由参与这条pathway 途径的蛋白的表达量或者蛋白的活性改变而引起。而通过芯片结果得到的是编码这些蛋白质的mRNA 表达量的变化。从mRNA 到蛋白表达还要经过microRNA 调控,翻译调控,翻译后修饰(如糖基化,磷酸化),蛋白运输等一系列的调控过程,mRNA 表达量和蛋白表达量之间往往不具有线性关系,因此mRNA 的改变不一定意味着蛋白表达量的改变。同时也应注意到,在某些pathway 中,如EGF/EGFR 通路,细胞可以在维持蛋白量不变的情况下,通过蛋白磷酸化程度的改变(调节蛋白的活性)来调节这条通路。所以芯片数据pathway 分析的结果需要有后期蛋白质功能实验的支持,如Western blot/ELISA,IHC(免疫组化),over expression(过表达),RNAi(RNA 干扰),knockout(基因敲除),trans gene(转基因)等。
- 基因网络分析 目的:根据文献,数据库和已知的pathway 寻找基因编码的蛋白之间的相互关系(不超过1000 个基因)。
GO的里面的relationship,B is a A是指B is a subtype of A,它定义的是这样从属的关系,显示的是I.还有一种relationship叫part of,显示的是P .B part of A的意思是B is a part of A,这主要是一种subunit和unit的关系。is a 有一种家族的关系,part of是一个部分。另外一类很重要的relationship是regulates,细分为subrelationship,一个是postive regulates,另一个是negative regulates,比如说regulate就是一个R显示的,positive 是绿色的R,negative是红色的R.在点奥存储仍然是用OBO和XML文件。 有了这些关系,就可以来做很多推断。比如说A is a B ,B is a C,那么就可以推断A is a C.如果A is a B,B is part of C,那么A is part of C,所有的逻辑关系都可以在计算机里定义。一旦定义,不管是什么基因,就可以做这些判断

Gene Ontology有一个Website,就是这个http://www.geneontology.org/ 里面一共定义了将近四万的概念 ,一共设计到了2000多个物种, 这里一个注释了57万多的基因,定义的关系包含7万5前多条关系。里面有一个AmiGO来辅助你浏览。
二、GO注释分类介绍
Gene ontology的注释主要有三大类:
- 一大类是雇佣大量的人看文献,把文献里的研究的基因分子功能录入到数据库里,所以庞大的Gene Ontology背后有很多的工作人员;
- 第二类是利用BLAST类似的方法把没有实验证据的基因通过序列相似性连接到与它相似功能已知的基因,但同样需要人来审核,所以叫anually-reviewed computional analysis evidence,测序的速度远远地超过了任何团队一个个来检验预测结果的速度;
- 在Gene Ontology还有一类完全自动生成的预测结果,没有人能看过,这一来的证据要弱一些。
通过人工审核的证据包括以下几大类,每一类都有明确的标号来标注

第二大类是人工审核过的计算结果,包括序列同源性如BLAST的方法,或者序列比对,

如果没有人工看过的,只是电子生产的,就会表标注IEA,也就是inffered from electroni annotation,

三、go的注释:
- 先对拼接出来的宏基因组数据预测orf
- 拿orf跟nr数据库做个balst比对
- 比对的结果用balst2go来注释
命令行:
#!/bin/bash
java -cp *:ext/*: es.blast2go.prog.B2GAnnotPipe -in /sam/syn/result/cluster9/nr/cluster9.xml -out /sam/syn/result/cluster9/go/cluster9 -prop b2gPipe.properties -v -annot -dat -img -ips ipsr -annex -goslim -wiki html_template.html
-prop 为参考设置
-v 把应用信息打在终端上
-annot 生成B2G注释文件(.annot)
-dat 生成B2G文件夹(.dat)
-fas 生成fasta文件,然后把BLAST和IPS结果加上去
-img 生成统计的图,必须有参数-dat才行
-ips
When a directory is provided here, the application will search for each sequence
a xml-files with the same name (.xml). The xml-file will be parsed and
the corresponding GO-terms found for domains/motifs will be added to the
annotations. (没看明白)
-wiki 基于给定的模板来生成wiki文件
-annex 运行Annex步骤
-goslim 把注释结果转化成Goslim格式
需要联网来生成goslim和GO-Graph总结的图
注释的结果可以用wego来图形化(貌似blast2go本来就具有图形化的功能吧)
四、其他
基因本地论的各种注释工具的汇总
- http://neurolex.org/wiki/Category:Resource:Gene_Ontology_Tools
- http://blog.sciencenet.cn/blog-402211-629121.html
参考资料
- 又听到丽萍老师的课了,爽啊,这些简单的概念我刚开始看文献的时候可费了一些经历了,受益颇多,期待下一节的课。
- 2014.1.15参考文献《大规模GO注释的生物信息学流程》对以上内容做了一些补充。
- 在水一方的博客 http://blog.sina.com.cn/s/blog_7295d993010131jx.html(超赞)
