【5.3.1】分子通路鉴定--KOBAS
KOBAS
官网:http://kobas.cbi.pku.edu.cn/program.inputForm.do?program=Annotate
把KO里面的条目的信息包括ID,对于的pathway等全部用python parse出来,用Mysql语句存入我们自己的数据库,剪了几个表格,包括KO的表格,pathways的表格和KO到pathway的表格,同时把KEGG数据库里相关的基因的序列和ID也存到数据库里面.支持genebanK GI, Entrez Gene ID, Ensemble Gene ID等等。有时候用IDmapping找不到几个通路,因此实现一个通过序列相似性进行mapping,基于的假设是同源基因在不同的物种里会参与类似的通路,这样就可以通过跨物种的比较借助研究得比较清楚的模式生物的基因。
sequencing mapping就是很简单的blast,每一个输入的序列和KEGG里的基因做一个blast比对,有两个cutoff值,evalue和rank,rank<6要求你的比对后的前5个基因只要有一个要有通路的注释。
评估一下序列比对的注释准确度:
precision = TP/(TP+FP)
coverage = TP/ N
如果标注严格的话,第一个值会很高
第二个值代表有多少正确的注释
把你的基因注释上去之后,到底哪个通路比较重要?
最直观的想法就是哪一个通路包含最多的我的基因,但这个是有点问题的,有一些分子通路就很大,即使是完全随机产生的一组基因,没有任何生物学意义,也会使大的通路覆盖的基因较多,任何实验都会有噪音,任何实验得到的基因都会有真的有假的,甚至有时候实验没有做好。我们需要软件用一定的方法把噪音和有意义的通路分开。
另一种策略是找最富集的通路的策略。
你研究的基因组里能注释的所有的基因叫做一个Backgroud.记为N.M个基因落在你感兴趣的pathway里面,你实验得到了比说说n个基因,那这些基因你通过注释发现,有m个基因落在这个通路,所有正个基因组有N个基因有M个基因落在这个通路里,你的实验里的n个基因有m个落在这个通路里。这个通路在你的实验结果里是不是很特别,就需要评估的事情。Null hypothesis,零假设,看到的是随机产生的,没有任何特别。我们用p-value来表示,它的含义是如果假设是真的,完全是随机看到的你的n个基因有m个落在这个通路里的概率是多少?如果这个概率非常小,那么组基因就可能是这个通路真的有关联。p-value越小,放错误的概率越小。一般p-value为0.01,0.05
超几何分布抽样的问题
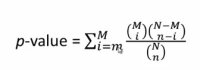
如果p-value为0.05,那么就有5%的概率看到是一个假阳性,这里我们只是在谈一个通路来做测验。KEGG里大概有360个通路,每一个通路都做超级和检验,每一次有5%的概率出错,一共进行360次,那出错的概率会很大。那如何评估最后的结果呢?我们需要算一个多假设检验的矫正,只要做了多次的statistical test,就要做多假设矫正。 一个办法就是算所有360个通路里,最后得到显著性的通路有一个或这更多假阳性的概率是多少,这是非常保守的,没有几个实验能多这么严格的检验。现在用的最多的是FDR矫正,它矫正的是false discovery rate,如果它的期望值小于0.05,那么大家就可以认为有可能是一个真实的生物学意义的结果。
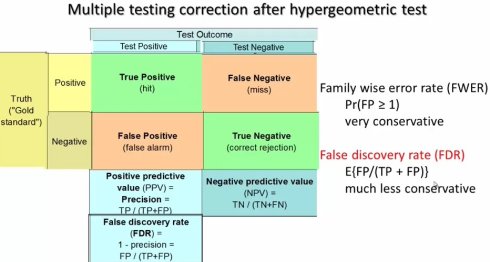
具体的操作进入KOBAS上的网页,分两步,一步是annotate,另一个是identify,也就是超几何检验。
ps:这个KOBAS主要两个功能,注释和富集分析,富集分析需要选择背景物种的基因组。
参考资料:
北大生物信息公开课
