【7.4.1】 MetaBGC--利用人类微生物组化学库的宏基因组策略
读取 reads 读数 reads
结构化摘要
介绍
人类微生物组与多种健康和疾病状况相关,但这些相关性背后的分子机制在很大程度上仍未得到探索。人类微生物组产生的生物活性小分子为探索这些机制提供了重要途径,因为它们通常介导重要的微生物-微生物和微生物-宿主相互作用。在细菌基因组中,小分子生物合成基因通常编码在称为生物合成基因簇 (BGC,biosynthetic gene clusters) 的不同簇中,这使科学家能够使用计算工具来识别它们并预测它们的产物。在这里,我们提出了一种混合策略,该策略使用计算和合成生物学工具来发现微生物组编码的小分子。
基本原理
以前从人类微生物组中发现小分子 BGC 的努力主要依赖于分析已测序的细菌分离株的基因组数据。尽管这种方法揭示了微生物组编码的 BGC 的巨大且大部分尚未开发的多样性,但它未能报告尚未培养或分离的人类微生物组成员(宏基因组测序数据中的大多数物种)的生物合成潜力。因此,我们寻求开发一种计算算法,直接在人类微生物组的复杂宏基因组测序数据中发现小分子BGC:生物合成基因簇的宏基因组标识符 ( MetaBGC)。
- 首先,用于识别感兴趣的生物合成酶的同源物的高性能概率模型专门用于复杂的宏基因组数据集 ( MetaBGC - Build )。
- 接下来,这些模型用于在单读取水平(MetaBGC - Identify)的人类微生物组的数千个宏基因组数据集中识别生物合成基因。
- 最后,在整个样本队列 ( MetaBGC - Quantify ) 中量化确定的生物合成读数,并根据其跨样本的丰度分布将其聚类到生物合成读数箱中 ( MetaBGC - Cluster)。
- 为了评估这种方法的实用性,我们使用它直接从人类微生物组的宏基因组测序数据中发现了 II 型聚酮化合物的 BGC,这是一类临床相关的小分子。
结果
我们应用了MetaBGC到来自西方(来自美国、西班牙和丹麦的受试者)和非西方(来自中国和斐济的受试者)人群以及每个主要人体部位(肠道、口腔、皮肤和阴道)。总的来说,
- 我们发现了 13 个可能编码 II 型聚酮化合物的完整 BGC;其中 8 种由人类微生物组的不同细菌分离株以菌株特异性方式编码,5 种无法分配给任何已测序的物种。II 型聚酮化合物 BGC 存在于三个主要人体部位,肠道、口腔和皮肤,其中至少有六个在宿主定植条件下转录并广泛分布在不同人群中(例如,46% 的健康受试者来自美国在其肠道、口腔或皮肤微生物组中编码至少一种 BGC)。
- 下一个,我们选择了两种已鉴定的 BGC 进行实验表征,一种来自口腔微生物组,另一种来自肠道微生物组。我们使用了一种合成生物学策略,其中宏基因组学发现的 BGC 被基因改造并在各种异源宿主中表达,而无需培养本地生产者。使用这种策略,我们成功地纯化并解析了五种新型 II 型聚酮化合物分子的结构,作为两种表征 BGC 的产物。
- 最后,我们展示了两个发现的分子对人类微生物群中的成员发挥强大的抗菌活性,这些成员与其生产者占据相同的生态位,这意味着可能在微生物-微生物竞争中发挥作用。
结论
我们开发了一种混合策略,它结合了计算和实验技术,用于直接从人类微生物组的复杂数据集中发现和表征小分子 BGC。使用这种策略,我们发现一类临床相关的分子,II 型聚酮化合物,在人类微生物组中广泛编码,并且人类微生物组衍生的聚酮化合物在结构和生物活性上与临床使用的相似。我们的方法通常适用于其他类别的小分子,可用于系统地揭示人类微生物组的化学潜力——这一目标对机械(mechanistic)微生物组探索和药物发现都有用。
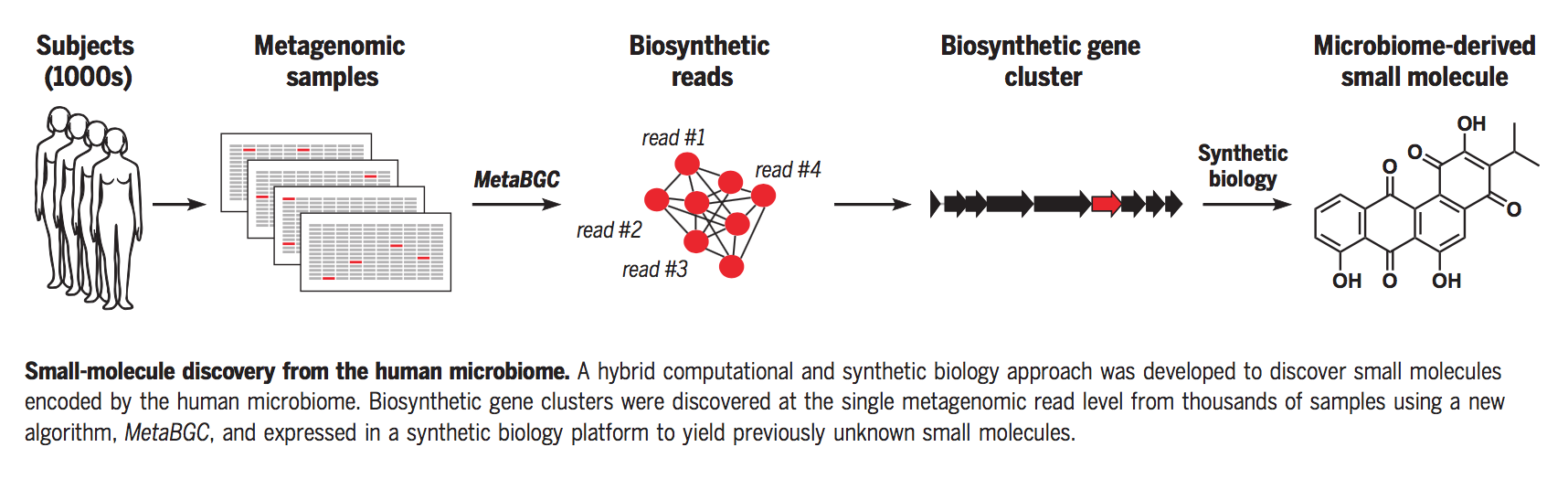
摘要
在确定微生物组对人体生理和疾病的影响方面已经取得了广泛的进展,但控制这些影响的潜在分子和机制在很大程度上仍未得到探索。在这里,我们将一种新的计算算法与合成生物学相结合,以访问直接编码在人类微生物组衍生的宏基因组测序数据中的生物活性小分子。我们发现临床使用的一类分子的成员在人类微生物组中广泛编码,并且它们对邻近微生物发挥有效的抗菌活性,这意味着可能在生态位竞争和宿主防御中发挥作用。
前言
人类微生物有数千细菌种类,改变在人体内的和个人之间的不同位点之间的组合物,并已与多种疾病相关(1 - 5)。最近,科学家们开始在机制层面上研究这些相关性,通常是通过识别和表征导致特定表型的微生物组衍生小分子 ( 6 , 7 )。这些分子可以直接介导其作用,通过靶向人细胞或受体(微生物-宿主相互作用)(8 - 14),或间接地通过影响微生物的其他成员在竞争或合作的方式(微生物微生物相互作用)(15 – 18 )。随着微生物组衍生小分子的重要性日益凸显,迫切需要开发系统方法来发现和表征它们。
我们之前采用了一种系统的方法来描述微生物组产生小分子的一般能力 ( 15 ),方法是鉴定最初从人类分离的数千种细菌菌株的基因组中编码的生物合成基因簇 (BGC) 库,并表征他们的次级产物结构和生物活性( 9 , 15)。尽管这种方法揭示了微生物组产生小分子的巨大且很大程度上尚未开发的能力,但它受到一个主要限制:它的起点依赖于分析来自易于培养和测序的人类微生物组分离物的组装基因组。因此,这种方法遗漏了在测序的临床样本中编码的 BGC,但没有遗漏先前分离的细菌基因组中的 BGC。由于三个主要原因,该限制值得注意。
- 首先,大多数大培养的努力集中于人类肠道微生物样品,特别是那些来自健康和西方人群,造成其他人的身体部位的参考基因组相对有限的代表性和从患病和非西方同伙(19 - 22)。相比之下,已经从所有人体部位 ( 1 , 2 )、几种人类疾病的队列 ( 3 – 5、23 – 25 ) 和非西方人群 ( 26、27 )产生了数千个宏基因组测序数据集。)。
- 其次,最近的宏基因组学分级研究表明成千上万的新菌种的人类微生物,较罕见,目前还没有培养或尚未测序成员(28 - 30)。这些新物种不仅扩展了人类微生物组的分类空间,而且其功能能力也超出了之前在培养分离株基因组中观察到的情况。
- 最后,BGC 通常在移动元件上编码为菌株特异性特征(15) 并捕获它们需要在单个物种中进行深度采样,这通常不是培养工作的目标。很明显,我们之前的方法仅依赖于从培养分离株的基因组中发现 BGC,在充分代表人类集体微生物组的生物合成潜力方面受到限制。
一种用于检测宏基因组数据中 BGC 的基于读取的算法 A read-based algorithm for the detection of BGCs in metagenomic data
意识到上述限制,我们着手开发一种算法,允许直接在人类微生物组衍生的宏基因组测序数据中识别小分子 BGC,而无需细菌培养或测序;我们称这个算法为MetaBGC(生物合成基因簇的宏基因组标识符,metagenomic identifier of biosynthetic gene clusters)。我们的算法需要满足三个主要标准:
- 能够从宏基因组测序数据中从头检测 BGC,而无需对样本中的细菌种类有任何先验知识,因此对测序分离株没有偏见;
- 能够在单个宏基因组读段 [~100 碱基对 (bp)] 上检测新的 BGC,因此不会偏向于通常在宏基因组组装中占主导地位的丰富或易于组装的物种;
- 在不牺牲灵敏度的情况下具有计算效率,因此可用于同时分析数千个宏基因组样本。
检测给定蛋白质家族同源物的一种简单快速的方法是通过轮廓隐藏马尔可夫模型 (pHMM,hidden Markov models) ( 31 , 32)。在 pHMM 中,在感兴趣的比对(训练集)的每个位置计算找到给定氨基酸、插入或缺失的概率,然后用于构建概率分布。然后根据新序列(搜索集)与该配置文件的匹配度对其进行评分。pHMM 通常由训练集中的全长蛋白质构建,而大多数基于 Illumina 的宏基因组读数长度约为 100 bp(约 33 个氨基酸)。当在搜索集中使用时,这些短序列会与它们各自 pHMM 的不同区域局部对齐。根据这些局部区域的序列复杂性和保守性,可以在给定的 pHMM 中获得不同的特异性和敏感性。所以,我们试图通过开发我们在此称为分段 pHMM (spHMM,segmented pHMMs) 的内容来调整 pHMM,以用于宏基因组应用。sphMMs 建立在 30 个氨基酸片段的对齐全长蛋白质同源物上,从而产生与搜索集中序列长度匹配的概率分布。
由于每个片段的尺寸小,spHMM 能够区分高度复杂的对齐蛋白质区域与序列中常见的重复或低复杂性区域,以及同源物中高度保守的区域与更复杂的区域。这种差异将导致模型的性能取决于它们的间隔(图 1A)。 使用合成数据集对模型进行评估,并消除性能不佳(具有高假阳性、高假阴性或低真阳性率)的 spHMM。 该算法的第一个模块被命名为 MetaBGC-Build。 下一步只选择高性能spHMMs,用于从临床样本中搜索宏基因组读数(MetaBGC-Identify)。
由选定的 spHMM 识别的读数(Reads)(得分高于定义的阈值)然后被认为是“生物合成的”,并传递到算法的第三个模块:MetaBGC -Quantify(图 1B)。在这个模块中,生物合成读数在整个队列的所有样本中被去复制和量化,并为所有样本的所有非冗余读数生成丰度矩阵。由于源自单个 BGC 的读数应该在宏基因组样本中具有均匀的覆盖范围,因此我们设计了一种聚类策略,以根据不同样本中的丰度特征生成已识别读数的“箱”。在这个策略中,我们使用 DBSCAN(基于密度的噪声应用空间聚类)(33) 基于成对 Pearson 相关距离将不同宏基因组样本中具有相似丰度分布的读数聚类到不同的箱中(参见补充材料)。第四个模块称为MetaBGC - Cluster。这一最终策略不仅减少了需要分析的命中总数,而且还为来自同一个 BGC 并最终在同一个 bin 中的读取进行有针对性的组装提供了机会。总之,我们基于 spHMM 的算法在单个宏基因组读取级别上识别、量化和聚类微生物组衍生的 BGC(图 1B)。

图 1 MetaBGC概述。 ( A ) MetaBGC 的第一步是开发特定于给定BGC类别的高性能 sphMMs ( MetaBGC - Build )。对感兴趣的蛋白质家族的同源物进行比对,并将比对分割成 30 个氨基酸的片段,并具有 10 个氨基酸的窗口偏移。spHMMs 是使用分段比对构建的,然后使用合成宏基因组进行评估,该宏基因组由主要为负背景和掺入正 BGC 组成。F1 分数≥0.5 的sphMMs构成MetaBGC -识别的基础,排除 F1 分数 <0.5 的人。( B ) MetaBGC剩下的三个步骤如下: (i)MetaBGC -使用 spHMM 从人类微生物组的复杂宏基因组数据集中识别检测生物合成读数 [如 (A) 中所述];(ii) 然后在所有样品中量化独特的(非冗余)生物合成读数(MetaBGC - Quantify),并为每个读数生成丰度曲线;(iii) 非冗余生物合成读数最终根据其丰度分布(MetaBGC - Cluster)进行聚类,以产生源自特定BGC 的读数“箱”。例如,从 10 个宏基因组样本中检测到红色或蓝色的生物合成读数,在相同的样本中进行量化,最后聚集以产生两个不同的 bin(一个红色和一个蓝色),它们源自bgc1或bgc2,分别。非生物合成读数为灰色。
测试用例MetaBGC:II型聚酮合酶BGCs
为了评估这种方法的实用性,我们专注于一类从未从人类微生物组成员中报道过的小分子 BGC:II 型聚酮化合物合酶 (TII-PKS BGC)。TII-PKS BGCs是在细菌基因组相对不常见,而且几乎总是编码小分子与感兴趣的生物学活性(包括临床上使用的抗癌药物多柔比星和临床上使用的抗生素药物四环素)(34,35)。为了首先测试是否可以在人类宏基因组测序数据中完全识别 TII-PKS BGC,我们对来自人类微生物组计划 1 (HMP-1-1) ( 1 ) 的 759 个样本进行了从头宏基因组组装) 并使用常见的 BGC 识别工具 antiSMASH 来检测组装支架中的 TII-PKS BGC > 5000 bp ( 36 )。使用这种策略,我们在三个身体部位(口腔、肠道和皮肤)(bgc1到bgc6;见下文)中鉴定了 6 个新的 TII-PKS BGC,这表明这类分子确实编码在人类衍生的宏基因组中,尽管没有已从人类微生物组的常见分离株中报道。受这一发现的启发,我们着手调整我们的MetaBGC直接在人源宏基因组测序数据中系统发现和量化 TII-PKS BGCs 的策略,在单读水平上,不依赖于宏基因组组装。这种策略将在很大程度上消除发现成功对编码生物的丰度或从复杂的宏基因组样本中正确组装其基因组的能力的偶然性。
四种必需酶普遍存在于 TII-PKS BGC 中:两种酮合酶(KSα 和 KSβ),分别负责聚酮链的延长和链长的测定;酰基载体蛋白 (ACP),也称为硫醇化结构域 (T),生长链通过硫酯键连接在其上;以及四种环化酶或芳香酶(OxyN、TcmN、TcmI 和 TcmJ 类型)中的至少一种,它们负责通过一系列羟醛缩合反应使聚酮链最终环化/芳香化(图 2A)(35 , 37)。尽管 KS 和 ACP 域存在于其他 BGC(例如,迭代、TI-PKS 和 TIII-PKS BGC)中,但环化酶域对 TII-PKS BGC 具有相对特异性,可用作使用MetaBGC识别它们的代理( 37 ) . 因此,我们比对了四种 TII-PKS 环化酶中每一种的选定的不同同源物,并以 30 个氨基酸的窗口大小和 10 个氨基酸的窗口偏移构建了所有间隔的 spHMM(参见补充材料,数据表 S1 ,以及图 S1 至 S5)。
评估MetaBGC的性能,我们将其应用于精心设计的合成宏基因组数据集。我们模拟了 140 个宏基因组样本,其中包含 42 个(低多样性)或 126 个(高多样性)人类微生物组来源的基因组,这些基因组不含 TII-PKS BGC。然后,我们从 10 个不同细菌基因组中加入了模拟读数,这些基因组包含总共 13 个 TII-PKS BGC,其中没有一个是 spHMM 训练集的一部分(参见补充材料,图 S6 和数据表 S2)。总体而言,该合成数据集(合成数据集 1)旨在模拟人类微生物组样本中预期的几种条件:给定 BGC 的丰度非常低(~1x 覆盖)、中等丰度(~10x 覆盖)、每个 BGC 的几个样本,并且每个样本没有 BGC(图 S7)。然后我们计算了 F1 分数(精度和召回率之间的谐波平均值)来单独评估所有四种环化酶的每个 spHMM。正如预期的那样,MetaBGC - Build揭示了每个比对的两种类型的 spHMM:高性能 spHMM(F1 分数 ≥0.5)和低性能 spHMM(F1 分数 <0.5)(图 2B)。在消除低性能模型并调整高性能模型的 spHMM 分数(图 S8)后,我们达到了MetaBGC -识别模块中使用的最终 40 个 spHMM 集。此外,正如预期的那样,这些模型检测到的给定环化酶的读数数量与含有相同环化酶的加标基因组的覆盖率呈正相关(图 S9)。总之,我们使用合成的宏基因组数据来评估 MetaBGC 中每个环化酶 spHMMs 的性能 并为后续步骤选择和调整性能最佳的。
为了评估MetaBGC的其余组件,我们将 40 个高性能模型中确定的“生物合成读数”置于量化和聚类模块中,并分析了生成的 bin 的组成。总共,MetaBGC产生了 11 个具有 >50 个非冗余生物合成读数的箱(范围 51 到 616 个读数,每个箱平均 278 个读数),其中 10 个是真阳性箱,其中最小的箱(箱 6、51 个读数)仅包含假阳性读数(图 S10)。总的来说,10 个真阳性箱包含来自所有 37 种加标环化酶的读数(100% 环化酶回收率)。接下来,我们调查了在 10 个真阳性 bin 中的每一个中代表了多少 BGC。10 个 bin 中的 7 个对应于完全源自七个非冗余 BGC 之一的读数,而其余三个 bin 分别包含来自两个 BGC 的读数(图 2C)。共享一个 bin 的每对 BGC 都在相同的尖峰基因组中编码,因此在宏基因组数据集中具有相同的覆盖率和表示,并通过以下方式聚集在一起MetaBGC -集群。总之,MetaBGC从所有加标BGC 的所有环化酶中恢复了读数,并将它们正确地聚类到它们的 BGC 和基因组特异性箱中。此外,MetaBGC在聚类后仅产生 4% 的假阳性读数,其中 40% 被合并到一个单一的、易于识别的假阳性箱中(图 2C)。
为了测试更广泛的 TII-PKS 阳性基因组覆盖分布是否会影响MetaBGC的性能,我们模拟了一个新的合成数据集(合成数据集 2),其中每个 TII-PKS 阳性和阴性基因组的比例在给定样本从对数正态分布中采样,然后归一化,使得比例之和等于 1(数据表 S2 和图 S11)。尽管在这个新数据集中对 TII-PKS 阳性基因组(0 到 >1000×)进行了广泛的覆盖,但MetaBGC以与以前相同的方式执行:总体而言,它产生了 12 个箱,每个箱中的非冗余生物合成读数 >50(10 个真阳性和两个假阳性),装箱后 95% 真阳性读数,100% 环化酶回收率,以及将环化酶读数 100% 正确分箱到相应的 BGC 和基因组中(图 S12 到 S14)。这些有希望的结果说明了MetaBGC在人源宏基因组中 TII-PKS BGC的鉴定、定量和聚类的有效性。

图 2 使用MetaBGC检测合成宏基因组样本中的 TII-PKS BGC。 (A)由编码两种酮合酶(KSα和KSβ)的基因组成的TII-PKS BGC的典型组织,分别负责增长的聚酮链的伸长和链长测定;硫醇化结构域 (T),生长链附着在该结构域上;和至少一种环化酶/芳香化酶(Cyc),负责线性链的环化。通过额外的剪裁反应,形成更精细的结构,例如临床使用的抗生素四环素。( B ) 四种类型的环化酶或芳香酶特别存在于 TII-PKS BGC 中,并被选为MetaBGC 的蛋白质家族. 对于四个蛋白质家族中的每一个,对应于 30 个氨基酸间隔的分段比对用于构建 spHMM。然后使用旨在模拟人类微生物组样本(合成数据集 1)中几种预期成分的合成宏基因组对每个 spHMM 进行评估。计算每个间隔(x轴)每个 spHMM的 F1 分数(y轴),这表示其准确性(同时考虑精度和召回率)。显示的值代表测试各种 spHMM 分数阈值后的最大 F1 分数(图 S8)。请注意,一些 spHMMs 的 F1 分数非常低,而其他人的 F1 分数很高(只有 F1 分数≥0.5 的spHMMs包含在MetaBGC -识别)。每个未分段的环化酶 pHMM 的 F1 分数以绿色显示。(C)堆叠条形图显示MetaBGC在应用于本研究中模拟的 140 个合成宏基因组(合成数据集 1)时产生的11 个 bin(每个 >50 个非冗余生物合成读数)。颜色表示生物合成读数属于每个箱内的环化酶类型。蓝色表示假阳性读数 (FP)。顶部的热图表示每个 bin 代表的 BGC 数量以及这些 BGC 源自的基因组数量。大多数 bin 代表单个 BGC,当两个 BGC 由同一个 bin 代表时,它们预计来自相同的加标基因组。合成数据集 2 的结果可以在图 2 中找到。S12 至 S14。
使用来自三个大型队列的宏基因组数据调整MetaBGC
在人类微生物组的模拟数据集上评估MetaBGC后,我们试图在临床衍生的宏基因组数据上调整其性能。我们将其应用于来自三项大型研究 [人类微生物组计划 (HMP-1-1 和 HMP-1-2) 和人类肠道联盟的宏基因组学 (MetaHIT) ( 1 , 2 , 38 )]。这些样本来自所有主要的人体部位(皮肤和呼吸道,293 个样本;肠道,872 个样本;阴道,215 个样本;口腔,1164 个样本),并且总共包含 1.09 × 10 ^11 个读数。通过比较MetaBGC根据国家生物技术信息中心 (NCBI) 非冗余蛋白质序列数据库的结果,我们进一步微调了 10 个 spHMM 分数截止值,以消除由宏基因组数据集中常见但最初未包含在我们的合成数据集中的基因组引起的假阳性命中,并淘汰了一个模型(见补充材料和数据表S1)。我们最终的、最优化的算法检测到 18 × 10^3读取(1.66 × 10 -7命中率),它们聚集成 19 个至少 10 个非冗余生物合成读取的箱(数据表 S3 到 S5)。
正如预期的那样,这些 bin 中有 6 个映射到我们最初的 6 个BGC集(bgc1到bgc6),13 个似乎源自新的(图 3和图S15 到 S17)。只有一个 bin,bin-W6,似乎包含两组具有不同丰度特征的读数(第一组平均在 72 个粪便样本中发现,而第二个仅在两个粪便样本中发现),它们很容易分成多个 bin W6a 和 W6b 使用更严格的聚类参数(图 3A、补充材料和数据表S5)。在 14 个新 bin 中丰度最高的 14 个选定样本上的各种组装方法提供了 10 个完整的 BGC 和四个部分的 BGC。在新的完整的 BGC 中,有六个显然是以前没有描述过的 TII-PKS BGC(图 4和数据表 S6)。antiSMASH无法识别六个新发现的 TII-PKS BGC中的四个(bgc9到bgc12)(36),即使在完全组装它们之后,也进一步突出了我们基于读取的发现方法的敏感性。对于所有完整的 TII-PKS BGC,每当 spHMM 在给定间隔内检测到环化酶读数时,该读数就会正确映射到完全组装的环化酶基因中的相同间隔(参见补充材料、数据表 S7 至 S10,以及图 S18)。剩下的六个完全组装的 bin 不代表 TII-PKS BGC,而是代表了几种新的含 OxyN 的 BGC 架构,以前没有特征性的功能(此处命名为:非聚酮化合物环化酶 BGC 或 NPC)(图 S15)
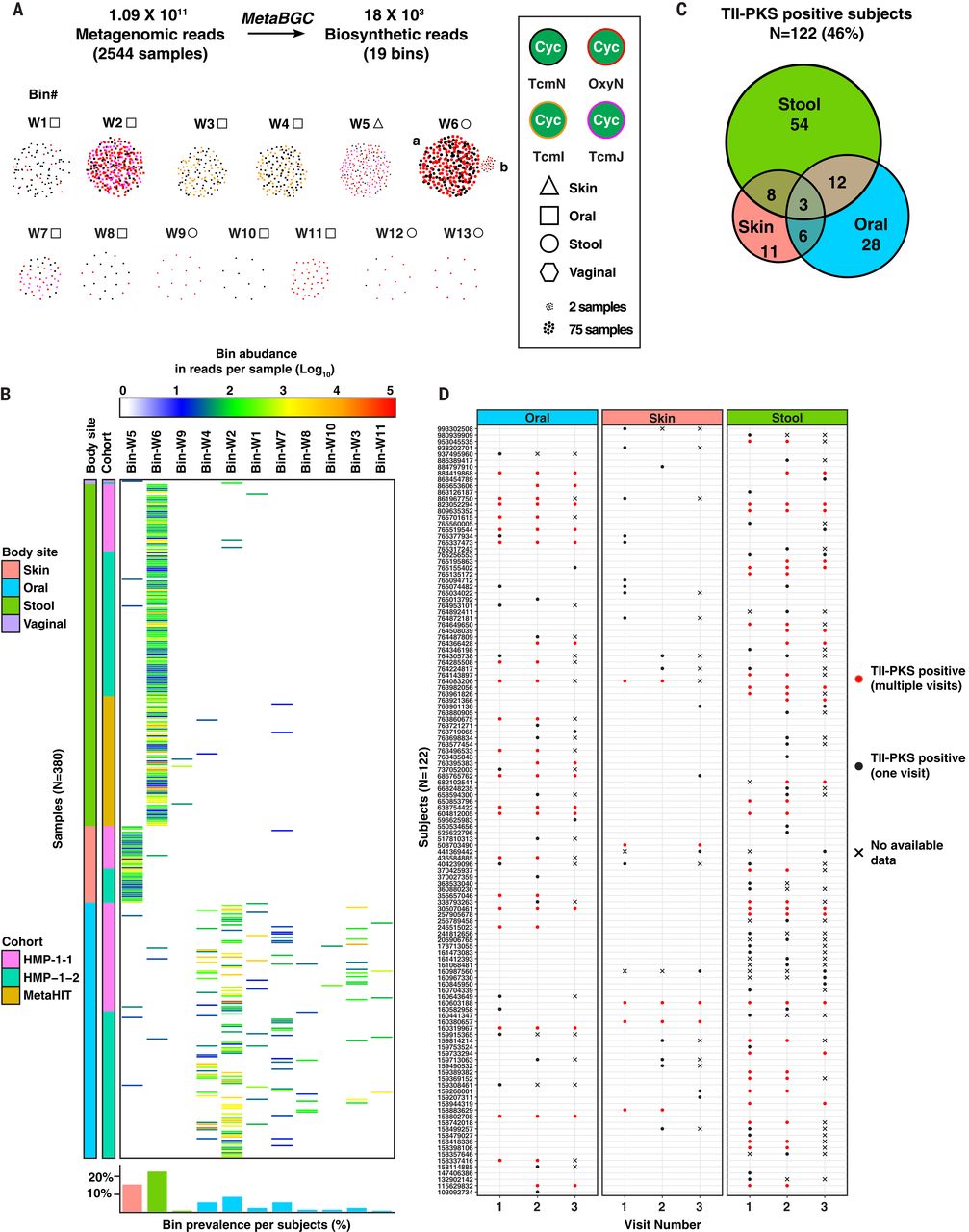
图 3 在来自三个大型队列的人类微生物组衍生宏基因组样本上测试MetaBGC。 ( A ) MetaBGC结果来自分析三个队列(HMP-1-1、HMP-1-2 和 MetaHIT)的 2544 个宏基因组样本。显示了与 TII-PKS BGC 对应的一系列 bins ( n = 13),其中每个点代表一个由MetaBGC检测到的非冗余生物合成读数。对于所有其他bins,请参见图。S16. 每个点的颜色、形状和大小表示每个读数所属的环化酶类型、每个读数起源的身体部位以及在右侧的键后面检测到每个读数的宏基因组样本数量。所有 bin 都包含来自一个 BGC 的生物合成读数。箱 W6a 和 W6b 使用精细聚类策略分离(见补充材料)。(乙) 热图指示 380 个宏基因组样本中对应于完整 TII-PKS BGC(数据表 S6)的 11 个 bin 中每个 bin 的丰度。有关所有其他 bin 的类似热图,请参见图。S17. bin 丰度计算为给定 bin 中所有生物合成读数的丰度总和(log 10),并在如上所示的渐变色标上指示(最小 bin 丰度为 10 个读数)。每个样本所属的身体部位和队列在左侧的颜色代码后指示。底部显示的条形图表示来自此处分析的三个队列的受试者中每个相应生物合成箱的流行率。在给定身体部位(嘴巴,219 名受试者;肠道,646 名受试者;皮肤,161 名受试者)的可用样本总数中显示了百分比。(C ) 维恩图,指示在一个、两个或三个身体部位(最小 bin 丰度为 10 个读数)中对完整 TII-PKS BGC 呈阳性的 HMP-1-1 和 HMP-1-2 受试者的数量。( D ) HMP-1-1 和 HMP-1-2 受试者的纵向分析。同一受试者最多 3 次访问的样本被视为 TII-PKS 阳性(点,最小 bin 丰度为 10 个读数)或 TII-PKS 阴性(无点)。红点表示受试者在随后的访问中具有相同的 TII-PKS bin(纵向一致性);黑点表示受试者仅在一次就诊时具有 TII-PKS 箱。X 表示没有可用宏基因组数据的访问。
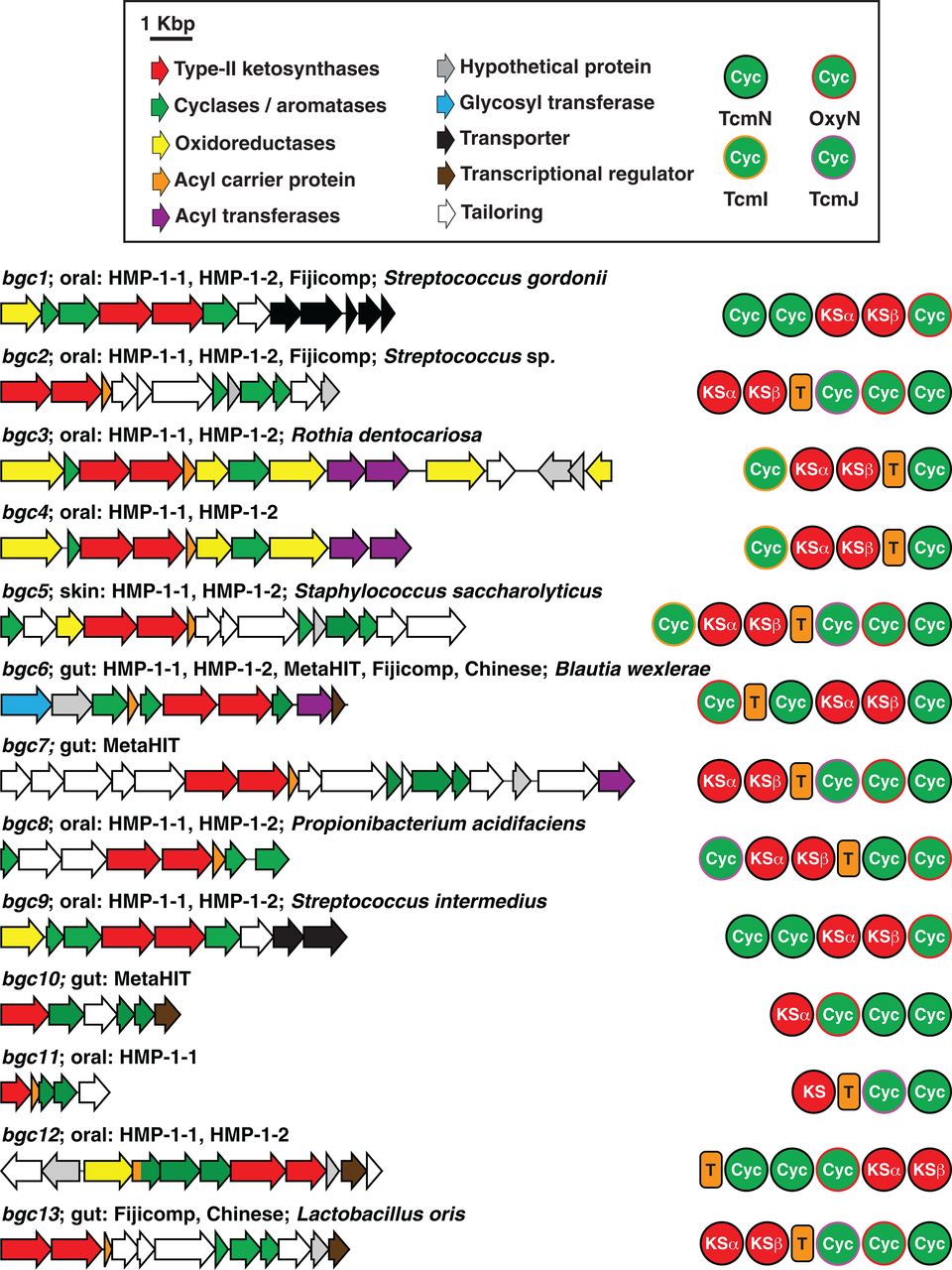
图 4 人源 TII-PKS BGC 的遗传组织。 每个 BGC 中的彩色箭头表示遵循顶部颜色代码的基因。典型的 TII-PKS 生物合成域(KSα、KSβ、T 和四种可能的 Cyc 类型)显示在每个 BGC 的右侧。还标明了发现每个 BGC 的主体部位、发现它的对象群组以及与其最匹配的细菌种类。请注意,几个 BGC 仅在宏基因组数据中发现,没有物种名称。更多详细信息,请参见数据表 S6。
TII-PKS BGC 在西方和非西方队列中都很普遍
然后,我们使用MetaBGC - Quantify来研究在 2544 个人类样本(660 名受试者)中产生完整BGC的所有 TII-PKS 箱的表示。每个样本的截止 bin 丰度为 10 个读数(参见补充材料),我们在任何身体部位的 122 名 HMP-1-2 和 HMP-1-2 受试者中检测到至少一个 TII-PKS BGC(46%)(共 265 个科目)。在这些 TII-PKS 阳性受试者中,93 名 (76.2%) 在口腔、肠道或皮肤微生物组中含有 TII-PKS-BGC,而 26 名 (21.3%) 和 3 名 (2.5%) 在两个或所有三个身体中含有它们位点,分别(图 3,B 和 C,无花果。S16 和 S17,以及数据表 S4)。通过分析来自同一受试者在不同访问中(相隔几个月收集)的纵向样本,我们发现 52%(口服)、52%(肠道)和 19%(皮肤)的受试者对给定的一次访问中的 TII-PKS 箱在随后的同一箱中呈阳性(图 3D和数据表 S11)。最后,在仅有肠道微生物组样本可用的 MetaHIT 队列中,395 名受试者中有 73 名 (18.5%) 携带至少一个 TII-PKS 箱(图 3B和数据表 S4)。
在对 HMP-1-1、HMP-1-2 和 MetaHIT 队列(来自美国、丹麦和西班牙的受试者)进行分析后,我们调查了 TII-PKS BGC 是否也在非西方队列中广泛存在。为了回答这个问题,我们分析了另外两个队列(Fijicomp,收集了来自斐济受试者的 434 个口腔和粪便样本,以及一个包含 225 个粪便样本的中国队列)(3, 26)。从 28 × 10 9 个总读数中,MetaBGC检测到 10 × 10 3 个读数(3.6 × 10 -7命中率),它们被聚集成 19 个 >10 个非冗余生物合成读数的 bin(图 S19 和 S20 以及数据表 S3 到 S5)。映射到先前在西方队列中发现的 TII-PKS BGC 和 NPC 的六个 bin;两个 bin 只包含假阳性读数,11 个似乎是新的。其中,11 个样本的组装使发现了 1 个完整和 4 个部分 TII-PKS BGC 和 5 个 NPC(图 4, 图。S15,和数据表 S6)。与西方队列相比,斐济和中国受试者分别有 15 (8%) 和 56 (25%) 的肠道微生物组中至少含有一种 TII-PKS BGC,而 38 (13%) 斐济受试者的肠道微生物组中含有至少一种 TII-PKS BGC。他们的肠道或口腔微生物组中存在 TII-PKS BGC,其中四个(1.4%)在两个身体部位均呈阳性(图 S19 和 S20 以及数据表 S4)。综上所述,这些结果表明 TII-PKS BGC 广泛存在于人体三个主要部位(口腔、肠道和皮肤)的不同人群的人体微生物组中,并且被人体的一致定植者所携带(稳定数月)。
因为MetaBGC通过量化仅源自其环化酶基因的宏基因组读数来推断给定 TII-PKS BGC 的丰度,我们试图进一步验证该推断的有效性。对于在给定队列中发现的所有完整 TII-PKS BGC,我们使用独立读取招募方法将同一队列中所有样本的宏基因组读取映射到 BGC 的整个长度,并研究了除四种类型的 TII-PKS 基因之外的 TII-PKS 基因是否可以检测到环化酶(见补充材料)。在超过 91% 的样本被MetaBGC视为阳性的情况下对于给定的 TII-PKS BGC,在样品中检测到映射到同一 BGC 内的环化酶和非环化酶基因的读数(数据表 S12 至 S16)。此外,在MetaBGC计算的 bin 丰度与以 RPKM(每千碱基对每百万测序读数)值表示的整个BGC的独立计算丰度之间观察到强烈的正相关:Pearson 相关系数为 0.85,p -值为 2.2 × 10 -16(数据表 S12 至 S16)。这些结果进一步验证了MetaBGC作为一种灵敏的、基于读取的算法,用于在人类微生物组的宏基因组样本中发现和量化 TII-PKS BGC。
TII-PKS BGC 由人类微生物组的不同成员编码,并在宿主定植条件下表达 TII-PKS BGCs are encoded by diverse members of the human microbiome and expressed under host colonization conditions
总的来说,我们直接从人类微生物组的宏基因组数据集中发现了 13 个完整的 TII-PKS BGC,每个都主要在一个身体部位(口腔、皮肤或肠道)中发现(图 3B和4)。为了更深入地了解具有编码这些 BGC 的基因组的人类微生物组成员,我们在 NCBI 和综合微生物基因组和微生物组 (IMG) ( 39 ) 中的参考基因组数据库中搜索了 13 个 BGC,并成功将其中 8 个映射到先前测序的细菌基因组(其余五个没有匹配;见补充材料)。我们发现 TII-PKS BGC 由一组不同的厚壁菌门(链球菌属、葡萄球菌属、Lactobacillus sp., and Blautia sp.) 和放线菌 ( Propionibacterium sp., and Rothia sp.)(图 4和数据表 S6)。即使在广泛采样的属中,映射的 BGC 也具有菌株特异性表示:bgc1和bgc9仅存在于约 3000 个测序的链球菌分离株中的一个菌株中,bgc5仅存在于 >5500 个测序的葡萄球菌分离株中的一个菌株中,而bgc13仅存在于约 1000 种已测序的乳酸杆菌中的一种隔离。这些结果表明,人类微生物组衍生的 TII-PKS BGC 由人类微生物组的已测序和尚未测序的成员以菌株特异性方式编码,进一步强调了直接从宏基因组测序数据中发现这些途径的重要性。
为了确定所发现的 TII-PKS BGC 是否确实在人体中表达,我们将来自不同人类口腔和粪便样本的公开可用的宏转录组数据映射到此处发现的 13 个完整的 TII-PKS BGC(参见补充材料)。总体而言,我们在至少两个不同的样本中观察到至少五种口服BGC (bgc1、bgc2、bgc3、bgc8和bgc9)和一种肠道 BGC(bgc6)的体内转录(图 S21 和数据表 S17)。这些结果表明 TII-PKS BGC 不仅在人类微生物组中编码,而且还在宿主定植条件下表达。
来自口腔和肠道微生物组的 TII-PKS BGC 的实验表征
为了确定人类微生物组衍生的 TII-PKS BGC 是否产生与其先前表征的环境细菌对应物相似的分子(结构和生物活性),我们选择了两种体内表达的 BGC 进行实验表征:口服的(bgc3)和一个肠道(bgc6)。由于在这项工作开始时尚未分离出含有bgc3的天然菌株,因此我们采用合成生物学策略对其进行表征。我们合成了针对链霉菌属中异源表达优化的bgc3编码序列,链霉菌属是一种广泛用于表达小分子 BGC(包括 TII-PKS BGC)的宿主 ( 40 )。bgc3在两个重叠片段中合成,具有驱动其两个潜在操纵子表达的强启动子(参见补充材料和图 5A)。然后通过在酿酒酵母中的转化相关重组将片段组装到大肠杆菌-链霉菌-酵母穿梭载体中,最后通过细菌结合整合到白色链霉菌染色体上的噬菌体附着位点(见补充材料)(41)。然后,我们比较重组的培养物的有机萃取物S.球菌:: bgc3与那些使用高效液相色谱与质谱联用 (HPLC-MS) 的空载体对照相比,揭示了几个bgc3特异性峰的存在(图 5B)。
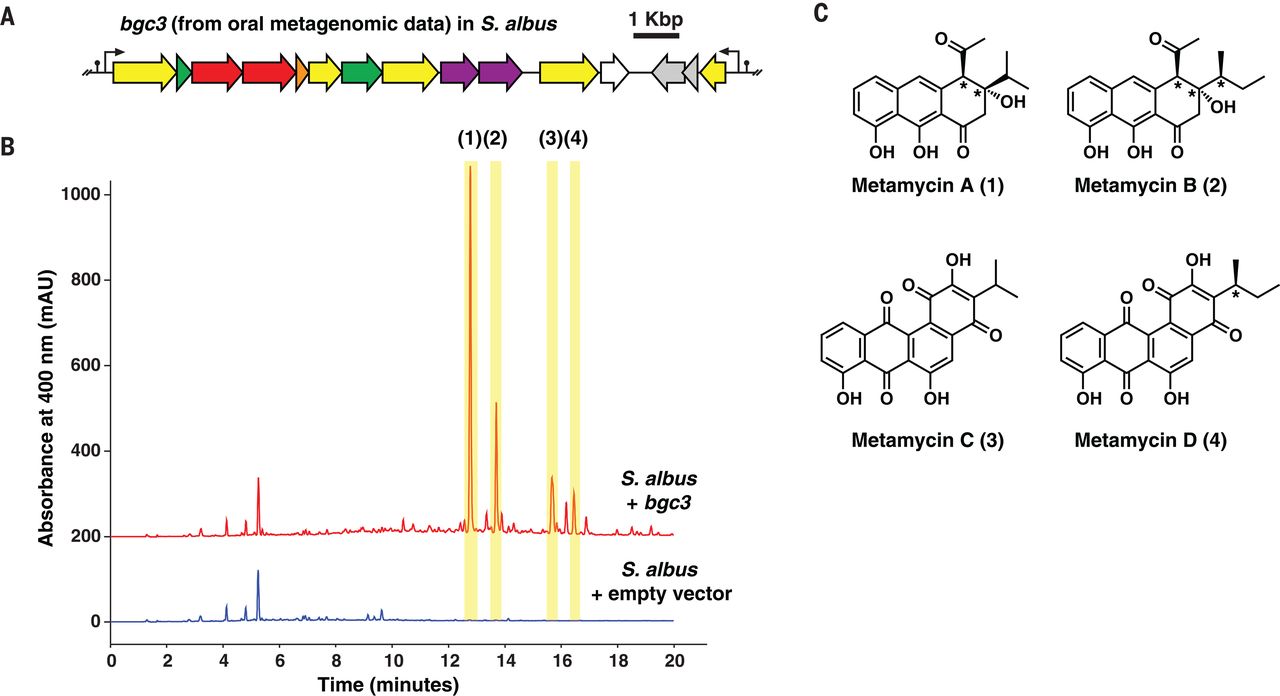
图 5 bgc3 的实验表征。 ( A )从人类口腔宏基因组数据中发现的bgc3的 DNA 序列是针对Streptomyces sp. 的密码子优化的。并合成、组装并克隆到异源宿主S. albus 中。终止子(黑点)和组成型启动子(黑色箭头)序列被设计为控制bgc3在S. albus 中的表达(见补充材料)。( B ) 来自S. albus培养物的化学提取物的 HPLC-MS 分析:: bgc3(红色)与来自S. albus的化学提取物的比较带有空向量控件(蓝色)。在400nm处的吸光度的HPLC色谱图显示两个样品,表明4个bgc3通过产生特异性峰S.球菌:: bgc3而不是控制(1至4,以黄色突出显示)。(Ç)metamycins A的分子结构,以d(1至4),的产品bgc3。星号表示手性碳的相对构型。
为了进一步表征bgc3产品,我们按比例增加的发酵S.黄鳝:: bgc3(27升),然后分离,纯化,并解决了四个新的II型聚酮化合物结构从它的有机提取物(化合物1至4,命名元霉素 A 至 D,分别;图 5 ),使用 HPLC-高分辨率串联质谱 (HPLC-HR-MS/MS)、一维和二维核磁共振 (NMR) 以及 X 射线晶体学的组合(参见补充文本,图 S22 至 S37,以及表 S1 和 S2)。元霉素 A ( 1 ) 和 B ( 2 ) 共享相同的芳香族三环骨架 (C16),但它们的起始单元在 C16(秒-丁基部分在2 中而不是在1中的异丙基部分)。元霉素 C ( 3 ) 和 D ( 4 ) 共享更长的四环芳族主链 (C18),同样在起始单元上不同。
我们表征的第二个 BGC 是bgc6(图 6A)。这种 BGC 对我们特别感兴趣,因为它在来自不同队列的人类粪便样本中的流行率各不相同,从斐济受试者的 7%,到丹麦或西班牙受试者的 17%,到中国受试者的 23%,最后到28% 的受试者来自美国(数据表 S4)。此外,bgc6编码在来自梭菌纲的肠道分离株Blautia wexlerae DSM 19850的基因组中。这是不寻常的,因为 TII-PKS BGC 很少在厌氧细菌中编码,并且已知只有两种厌氧菌会产生芳香族聚酮化合物 ( 42 – 44 )。表征bgc6 的乘积, 我们从B. wexlerae DSM 19850的基因组 DNA 中扩增其编码序列,在强组成型启动子下将其克隆到E.coli - Bacillus subtilis穿梭载体中,并通过自然方式将其整合到B. subtilis 168-sfp的基因组中转化和双交叉同源重组产生枯草芽孢杆菌-168- sfp :: bgc6(见补充材料)。在这种情况下选择枯草芽孢杆菌是因为它与B. wexlerae (Firmicutes)属于同一门,其基因组 DNA 中的鸟嘌呤胞嘧啶 (GC) 含量与B. wexlerae 相似(GC 含量在B. wexlerae 中为 41.5%,在B. subtilis -168 中为43.5% ),并且之前已被用作表达 I 型聚酮化合物(例如 6-脱氧赤藓内酯 B)的异源宿主,因此不应仅限于主要的聚酮化合物底物,例如醋酸盐和丙二酸酯 ( 45 )。
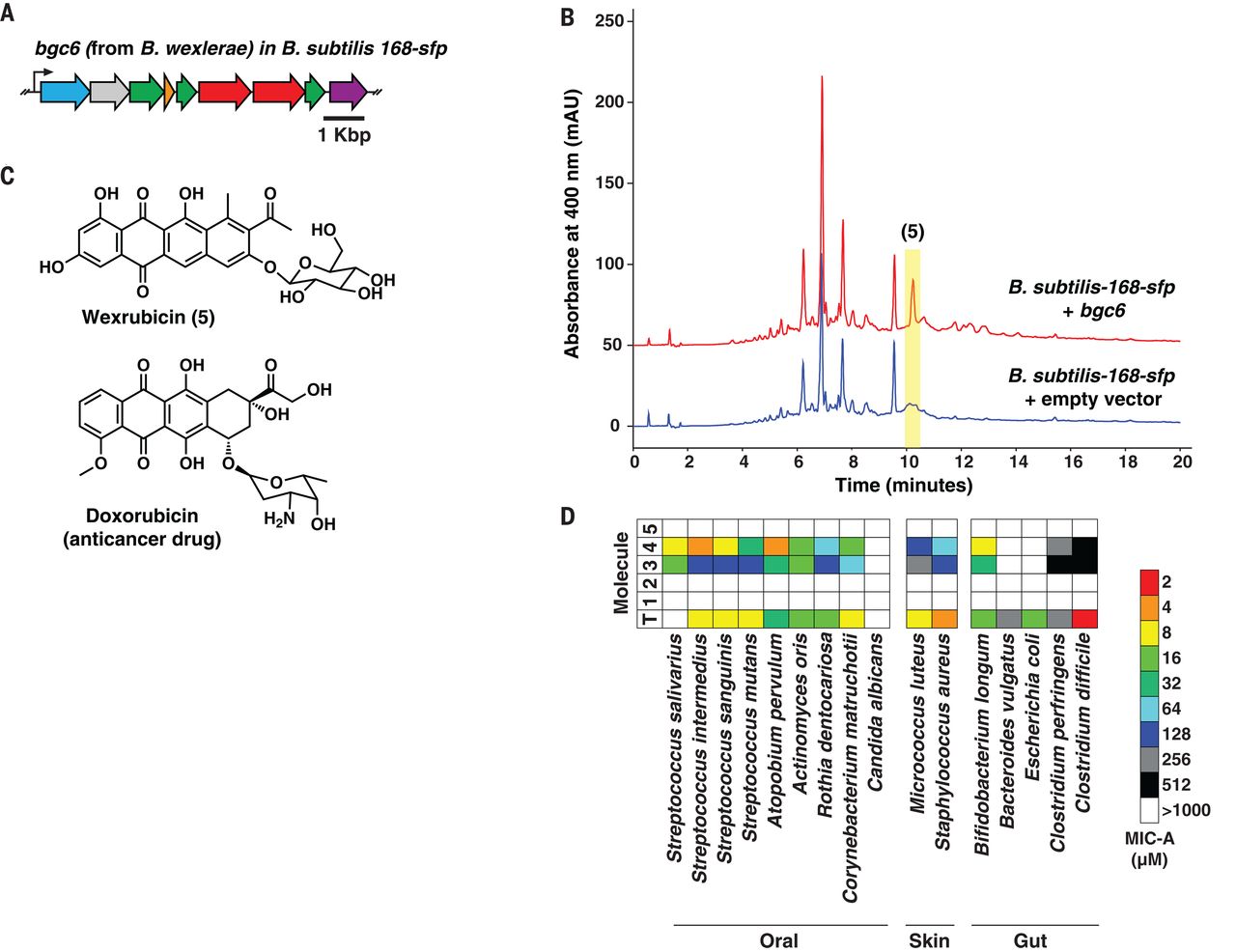
图 6 bgc6 的实验表征。 ( A ) bgc6的 DNA 序列是从肠道分离株B. wexlerae DSM 19850 中克隆到异源宿主B. subtilis - 168 - sfp 中的。黑色箭头表示组成型启动子,该启动子被设计用于控制bgc6在枯草芽孢杆菌中的表达- 168 - sfp(参见补充材料)。( B ) 对来自枯草杆菌培养物的化学提取物的 HPLC-MS 分析- 168 - sfp :: bgc6(红色)与来自枯草杆菌的化学提取物相比-168 -带有空矢量控件(蓝色)的sfp。显示了两个样品在 400 nm 吸光度处的 HPLC 色谱图,表明由枯草芽孢杆菌产生的单个bgc6特异性峰- 168 - sfp :: bgc6而不是对照(5,以黄色突出显示)。( C ) bgc6的产物威柔比星( 5 )和相关抗癌药物阿霉素的分子结构。( D ) 热图显示了元霉素 A 至 D ( 1至4 ) 和韦柔比星 ( 5 )的抗菌活性)针对一组具有代表性的人类微生物组的口腔、皮肤和肠道分离物。活性以微摩尔为单位测量,作为琼脂上的最小抑制浓度 (MIC-A)。以相同的方式测试抗生素四环素(T),并将其活性与新发现的聚酮化合物的活性进行比较。请注意,只有metamycin C ( 3 ) 和metamycin D ( 4 ) 对几种分离株具有抗菌活性,在某些情况下,metamycin D 的活性与四环素相似。
然后我们使用 HPLC-MS比较了枯草芽孢杆菌- 168 - sfp :: bgc6和空载体对照的有机提取物,揭示了单个bgc6特异性峰(图 6B)。接下来,我们放大了枯草芽孢杆菌-168- sfp :: bgc6培养物 (31L),然后分离、纯化并阐明了单个新分子(化合物5,命名为wexrubicin)使用HPLC-HR-MS/MS和NMR的组合(参见补充文本、图S38至S44和表S3)。Wexrubicin 具有与 C4 处的 β-葡萄糖部分连接的四环蒽环类环系统 (C21),这与bgc6 中编码的糖基转移酶一致(图 6C)。
人类微生物组中编码的 II 型聚酮化合物的生物学活性
1到5的发现不仅表明人类微生物组编码以前未描述的 II 型聚酮化合物,而且其中一些分子在很大程度上类似于临床使用的药物。例如,Wexrubicin ( 5 ) 属于蒽环类聚酮化合物,包括临床上使用的抗癌药物阿霉素和达努比星,以及抗肿瘤抗生素分子四烯霉素和埃罗霉素 ( 46 , 47 )。相反,元霉素 A 和 B ( 1 , 2 ) 以及 C 和 D ( 3 , 4 ) 类似于先前从S. pseudovenezuelae 中分离出的抗生素 setomimycin和 oviedomycin分别来自抗生素链球菌( 48 , 49 )。为了确定微生物组衍生的聚酮化合物是否发挥与其密切相关的衍生物相同的生物活性,我们在两种类型的测定中对其进行了测试:对哺乳动物细胞系的细胞毒性和对选定细菌和真菌的抗菌活性(参见补充材料)。与作为阳性对照的多柔比星相比,没有一种测试分子对 HeLa 细胞系显示出显着的细胞毒性(图 S45)。
然而,在抗菌试验中,metamycins C 和 D ( 3 , 4 ) 对几种革兰氏阳性菌表现出很强的抑制活性,在某些情况下,其抑制活性与临床使用的抗生素四环素相似。这些分子对链球菌、Atopobium、放线菌、Rothia和棒状杆菌的口服分离株最有效。(图6D )。作为口服 BGC ( bgc3 )的产物,这些结果表明,metamycins C 和 D ( 3 , 4) 可能在口腔内的生态位竞争或针对病原体的宿主保护中发挥作用。这与bgc3在早期生物膜形成期间在人龈上斑块样品中表达的事实一致,如宏转录组学分析所确定的(图 S21 和数据表 S17)。由于未检测到元霉素 A 和 B ( 1 , 2 ) 或威柔比星 ( 5 ) 的细胞毒性或抗菌活性,因此需要进一步研究以深入了解它们的生物学作用。
讨论
尽管在记录和表征由人类相关细菌编码的各种小分子方面付出了大量努力,但之前没有从人类微生物组中描述过芳香族聚酮化合物。在这里,我们使用了一种新的计算策略直接从人类微生物组衍生的宏基因组测序数据中发现小分子 BGC,揭示了人类微生物组中编码这种相对稀有的分子的 BGC 的广泛分布(约 50%来自美国的科目至少携带一个此类的 BGC)。然后,我们将此计算机方法与合成生物学策略相结合,以异源表达已识别的 BGC 并直接发现它们的化学产品。在此处发现的 13 个 BGC 中的两个产物中观察到的结构多样性,并且它们在结构或生物活性方面与临床使用的药物相似,这一事实显然激发了对该类药物的进一步功能研究。这些研究不仅将作为在分子水平上阐明微生物组-宿主相互作用的重要途径,而且还将作为从人体内发现药物的前所未有的资源。
我们的计算策略依赖于重新利用和定制已建立的概率模型,以与短读长宏基因组数据一起使用,从而实现目标蛋白质家族的高灵敏度和高特异性检测。我们在本研究中关注的不是一个而是四个不同的蛋白质家族作为原理证明(TII-PKS 环化酶和芳香化酶),这证明了我们方法的通用性。相同的方法可以很容易地适用于任何感兴趣的蛋白质家族,包括特定于其他类型 BGC 的蛋白质家族。为了说明这一点,我们将相同的策略应用于两个新的蛋白质家族,这些蛋白质家族特定于 BGC,这些蛋白质家族编码另外两种类型的小分子(铁载体和羊毛脂肽)。在这两种情况下,我们都成功地分离了低性能和高性能 sphMM,并使用了以下四个模块MetaBGC使用合成数据集 2(参见补充材料、数据表 S18 和图 S46 至 S49)。
MetaBGC的性能和局限性取决于几个因素。
- 首先,要用作给定 BGC 的发现和量化方法,重要的是要为感兴趣的 BGC 中特异且普遍存在的蛋白质家族构建 spHMM。
- 其次,在分割之前需要生成一个相对高性能、全尺寸的 pHMM,这反过来又依赖于足够数量的可用性和感兴趣的蛋白质家族的已测序同源物的正确对齐。
- 第三,需要生成精心设计的合成数据集,以尽可能多地反映感兴趣的真实宏基因组数据集的预期复杂性,并评估 spHMMs 并调整其分数截断值。
- 最后,需要根据具体的研究目标优化几个灵活的参数,包括 spHMM 片段的大小(取决于搜索数据的读取长度)、F1 和 spHMM 分数截止(取决于发现的保守程度) 目标是),定义新 bin 的非冗余生物合成读数的最小数量,定义宏基因组样本中给定 bin 的“存在”或“不存在”的最小 bin 丰度(取决于研究设计的敏感程度) ),最后是聚类步骤中使用的最小 Pearson 相关距离,它决定了分箱的严格程度。 我们在当前的研究中为这些参数提供了示例设置,可用作新应用程序的起点,并根据需要进一步优化。
总之,我们提出了一种通用策略,可用于快速分析来自大型临床队列的宏基因组数据集,并优先考虑候选 BGC 进行实验表征。更广泛地说,我们将其视为揭示人类微生物组编码的化学库的系统策略,这是了解其在人类健康和疾病中的作用所急需的一步。
实验方法
略
参考资料
- Science,2019. A metagenomic strategy for harnessing the chemical repertoire of the human microbiome. https://science.sciencemag.org/content/366/6471/eaax9176
