【1.1.2】jellyfish--kmers统计
JELLYFISH是CBCB(Center for Bioinformatics and Computational Biology)的Guillaume Marçais 和 Carl Kingsford 研发的一款计数 DNA 的 k-mers 的软件。该软件运用 Hash 表来存储数据,同时能多线程运行,速度快,内存消耗小。该软件只能运行在64位的Linux系统下。其文章于2011年发表在杂志 Bioinformatics 上。
Jellyfish是CBCB(Center for Bioinformatics and Computational Biology)的Guillaume Marçais 和Carl Kingsford 研发的一款计数DNA 的 k-mers 的软件。该软件运用 Hash 表来存储数据,同时能多线程运行,速度快,内存消耗小。该软件只能运行在64位的Linux系统下。其文章于2011年发表在杂志Bioinformatics 上。在其cbcb官网上有pdf版本使用手册,有详细介绍。jellyfish的功能有:kmer计数;融合二进制的Hash结果;统计Hash结果;通过Hash结果来画直方图;将Hash结果输出成文本格式;查询指定k-mer的数目。
一、背景介绍
在得到下机数据*.fq.gz后,先通过jellyfish计算k-mer分布频率,然后通过Genomescope2估计基因组大小、重复和杂合度。首先,我们需要先知道什么叫K-mer
1.1 K-mer定义:
从一段连续序列中迭代地选取长度为K个碱基的序列,若每条序列的长度为L,则K-mer长度为K,那么可以得到L-K+1个K-mer,后面我取K=21来进行分析(针对长度为K的K-mer,对于A,T,C,G四种碱基类型,一共能产生的K-mer种类数为4的K次幂个,选择K=21是为了产生足够多的K-mer种类数去覆盖整个基因组,当然,你也可以设置不同的K值进行探索,21是比较常用的)。
此外,针对一些物种可能会有较高的杂合率及多倍性,K-mer分布图会有不同的特征,比如:杂合K-mer,重复K-mer,需要我们进一步了解掌握。

对于不同的基因组杂合度,Kmer分布如下:
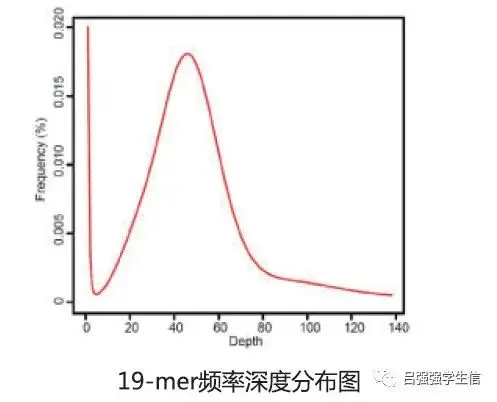
图中有一个明显的主峰,没有其它峰值,根据Kmer分布图,可初步判断该物种基因组为简单基因组。
高重复基因组
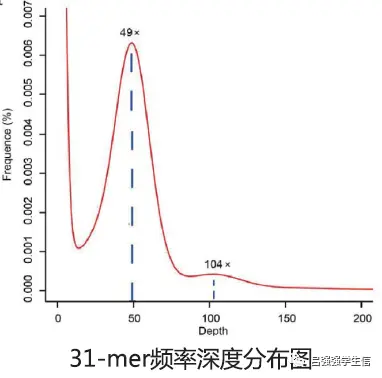
图中在49x深度和104x深度位置分别有1个峰,其中49x的位置为主峰即基因组正常期望深度,104x位置为重复峰位置。根据kmer 分布图,可初步判断该物种基因组为高重复基因组。
1.2. 基因组大小预估
我们需要了解K-mer深度分布曲线含义:K-mer深度—K-mer深度频率。从所有测序Reads中逐碱基取K-mer,并统计K-mer深度频数分布,从而获得K-mer深度分布曲线。根据曲线获得K-mer深度估计值,用于估计基因组大小。

基因组大小=(基因切割的Kmer数目)/(主峰深度)
K-mer分析适用于分析唯一主峰区域所占比例较大的基因组,当基因组杂合非常高或者重复序列比例非常大时,其影响可能导致无法通过K-mer分析正确估计基因组大小。将K-mer深度等于1的情况认为是错误情况,计算错误率,并用于修正基因组大小。
二、安装
wget https://github.com/gmarcais/Jellyfish/releases/download/v2.3.0/jellyfish-2.3.0.tar.gz
tar -xzvf jellyfish-2.3.0.tar.gz
cd jellyfish-2.3.0
./configure # --prefix=$PWD
make -j 4
make install
安装位置:/usr/local/include/jellyfish-2.3.0/jellyfish
更多用法,整理来自:https://www.csdn.net/tags/MtTaEgzsNTk2NDE2LWJsb2cO0O0O.html
三、用法
3.1 Jellyfish count
我们要用的是count 命令来进行K-mer计数,使用fastq文件在默认参数上和fasta文件没有区别,生成的hash结果为*.jf为后缀的二进制文件。
以count基本命令为例:
jellyfish count -m 21 -s 100M -t 10 -C reads.fasta
参数含义可以通过jellyfish count –help来进行理解,(大部分参数默认就可以):
其中我觉得可以根据自己需求进行设置的有:
-m使用的k-mer的长度。如果基因组大小为G,则k-mer长度选择为: k ~= log(200G)/log(4);
-s Hash 的大小,最好设置的值大于总的独特的(distinct)k-mer数,这样生成的文件只有一个。若该值不够大,则会生成多个hash文件,以数字区分文件名。如果基因组大小为G,每个reads有一个错误,总共有n条reads,则该值可以设置为[(G + n)/0.8],该值识别M 和 G;
-t |--threads=<num> default: 1 使用的CPU线程数;
-o |--output=<string> default:mer_counts 输出的结果文件前缀;
-c |--counter-len=<num> default:7, k-mer的计数结果所占的比特数,默认支持的最大数字是2^7=128。对于基因组测序覆盖度为N,则要使设置的该值要大于N。该值越大,消耗内存越大;
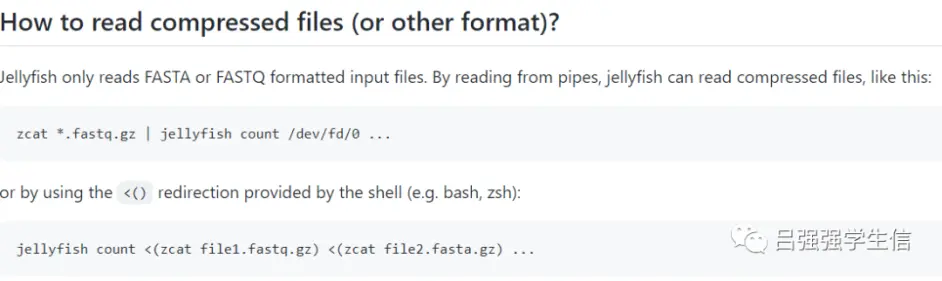
值得注意的是,我们的下机数据通常都是*fq.gz压缩文件,这个时候通过zcat命令及管道符“|”进行计算,一定要保留/dev/fd/0 , 这个是进程输入标志,也就是代表管道符前边的结果传递,其它参照上述参数设置。
3.2 stats命令对hash结果进行统计
k-mer的结果以hash的二进制文件结果给出,需要统计只出现过一次的kmer数;特异的k-mer数目;总的k-mer数;出现了最多的k-mer的数目.
jellyfish stats *.jf -o kmer_count
示例结果cat kmer_count:
Unique: 106239 #只出现过一次的k-mer的数目
Distinct: 153852 #特异性的k-mer数目
Total: 2974220 #总的k-mer数目
Max_count: 1085 #同一个k-mer出现最多的数目 作者:笨笨熊爱吃肉 https://www.bilibili.com/read/cv16360242 出处:bilibili
3.3 histo输出k-mer频率直方图数据,如:
对k-mer的计数结果有个直观的认识,则需要统计出现了x(x=1,2,3…)次的kmer的数目y,以x,y为横纵坐标画出直方图。使用 histo 命令能给出 x 和 y 对应的值,将结果默认输出到标准输出。
jellyfish histo mer_counts.jf > mer_counts.histo
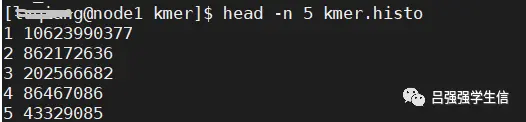
得到*.histo后缀的文件后,就可以通过Genomescope2进行K-mer分布结果的可视化作图了,并得到预估的基因组大小、杂合率、重复序列比例。
3.4 Counting k-mers in sequencing reads
jellyfish count
#常用参数
-m #counting kmer occurrence的过程中,所使用的kmer数。
-t #counting kmer过程中,所使用的线程数。
-s #预估在countin kmer过程中,有多少kmer会被储存到hash文件中。
-C #正、反义链都进行计算。
-F #同时打开文件的数量。比如,NCBI上下载的SRA数据,再经过fastq-dump后,可能是mate-pair或者pair-end的数据类型,前者为3个fastq,后者为2个,因此就设置对应的数值。
-o #prefix的设置。
-Q #设置一个阈值,任何质量值小于该值的碱基会被改为N。
Input为测序数据时,用k-mer遍历,但是该kmer是来自于正义链还是反义链都是未知的。但是从本质上来说,k-mer是等价于它的反义k-mer的。因此此时我们需要设置-C参数。该参数只将canonical kmer保存,但是频率为该k-mer出现的频率与其反义k-mer的频率。
Jellyfish-2与Jellyfish-1不同的点是,Jellyfish中的-s参数只是一个估计值。如果给定的size太小,而不能容纳所有kmer,那么结果hash文件的大小就会自动扩增或者会暂时将结果文件写入到磁盘中,最后自动整合在一起。因此Jellyfish-1中的merge命令就不需要了。
Counting higher-frequency kmers
在用Jellyfish计算测序数据中出现的kmer频率的过程中,可以将kmer分为2种类型:
- low-frequenc kmers
- high-frequency kmers
在 low frequency kmers 中,即测序数据中出现次数少的kmer中,只出现一次的kmer,极有可能是因为random sequencing error所产生的kmer,同样也会被储存到hash文件中。 所以应该如何获得高频k-mer呢?
有2种方法:
- one pass method
- two pass method
示例:
#one pass method
jellyfish count -m 25 -s 3G --bf-size 100G -t 16 homo_sapiens.fa
在counting all k-mers的基础上,加入–bf-size参数,会在jellyfish count运行的时候,先将得到的k-mers用Bloom filter进行过滤,然后将至少出现过1次的kmer添加到结果hash文件中。
–bf-size的设定数值应当是counting kmer结果预期产生的k-mers的数量。
-size的设定应当是counting过程中,出现次数大于1的k-mers的数量。
在Jellyfish-2的使用手册中,human的推荐参数设置为25-mer和30×的测序深度。
#two pass method
jellyfish bc -m 25 -s 100G -t 16 -o homo_sapiens.bc homo_sapiens.fa
jellyfish count -m 25 -s 3G -t 16 --bc homo_sapiens.bc homo_sapiens.fa
two pass的流程中,jellyfis bc会产生一个Bloom counter,之后Bloom counter会传递给jellyfish count,同时只有在Bloom counter过程中出现过2次或2次以上的k-mers会被保存到结果hash文件中。
参考资料
