【3.0】宏基因组聚类-binning-classification
在分析宏基因组数据的过程中,聚类(binning or classification)就是将reads或者contigs根据他们的性质分配到不同的OUT中,聚类的方法主要有三种:基于序列组成特征,序列比对,或者是二者之和。
宏基因组样品测序后包含来自不同物种大量的reads,例如对于1g土壤样品中,有将近18000种物种,每一个物种都会有自己的基因组【1】.宏基因组研究是将环境样品中所有的微生物的DNA给提出来,然后一起测序。大多数情况下,通过测序获得的不完整的序列很难聚类出独立的基因组【2】。因此,聚类技术知识尽力去通过识别reads或contigs特殊的地方以及跟其他序列相近的地方来分出OUT的分类【3】.
最开始不同物种混在一起的DNA通过特殊基因来鉴别分离开【4】【5】.这些标记基因是来自已知物种的克隆基因,因此,一旦这些基因出现在我们宏基因组的序列中,我们就可以将该序列归为我们已知的那个物种的OUT。这个方法的问题就是仅仅只有少量片段的序列包含标记基因,这样导致大多数的数据不能归类。
现在聚类的技术结合了两种方法,一方面使用先前样品可用的信息(比对),一方面结合测序以及物种所特有的信息(GC)【6】。基于样品的复杂性和多样性,有的情况下我们可以将序列归到中的水平上,然而有的仅仅只能归到非常宽泛的分类组上。
算法:
Mande et al., (2012)
【7】写了一篇综述关于针对宏基因组测序后的数据不同聚类方法的前提,方法,优点,缺点和挑战。见下面:
聚类分析存在3个重要的挑战:
- 相关数据库中缺乏相应的序列;
- 测序错误以及测序段;计算功能,算法要求高。
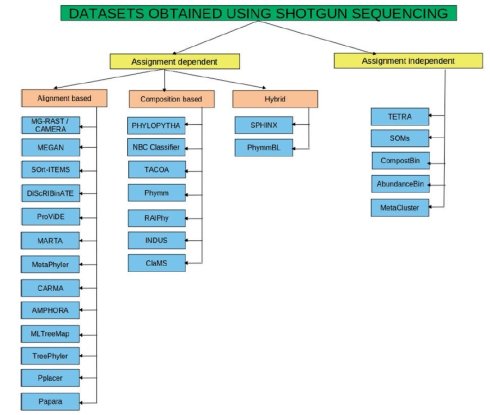
从上图我们可以知道,聚类分析的方法可以分为两类,一种是基于已知信息的比较(taxonomy depdent),一种不基于已知信息来比较(taxnomy indepdent);而高通量测序的数据中还包含物种丰度的信息,也可以用来聚类而taxonomy depdent又可以分为三类,alignment-based,composition-based,hybrid。alignment-based是跟已知数据库比对或者是跟已知序列建立的HMM模型比对,这种方法缺点是1,计算量很大,2因为大量物种未知,如果仅凭最好的比对值就认定为同一物种的的话,容易错误分类;composition-based是基于已知序列的序列组成成分,然后根据贝叶斯,支持向量机来建立序列模型,基于的原则是一个基因组,一个基因组组成,比如说根据GC,codon usage,oligonucleotide usage等,优点是快,但对序列长度要求更高;hybrid两者兼顾。 而taxnomy indepdent仅仅是测序出来的数据之间的比较来聚类,不依据已有的任何数据库中序列的信息。
TETRA
TETRA是利用基因组片段四核苷酸多态性的统计聚类方式【8】.在DNA中有4种碱基,因此4个连续的碱基就有256种情况,而这4个碱基的片段我们叫做tetramers(四聚物)。TETRA就是计算出一个给定的序列的每种四聚物的概率,然后通过这些概率来计算出z-scores,俩俩序列之间通过比较向量化后的z-scores来判断是否可以归为一类,一般z-scores相似度高的,我们就可以认为他为一类。
Phylopythia
Phylopythia 是由IBM实验室发明的一种基于支持向量机,通过之前已知序列的已知信息来来判断宏基因组中某些片段应该归为哪一类【5】。
SOrt-ITEMS
SOrt-ITEMS【9】是由印度的Tata Consultancy Services (TCS) Ltd实验室开发的基于序列比对的算法工具。胡勇需要将输入的宏基因组序列跟nr蛋白数据库通过blastx比对来寻找相似性,生成的blastx结果作为SOrt-ITEMS的输入数据。这个方法先是通过设定阈值来进行blast比对,这样来为这些reads鉴定出一个适当的分类地位.基于直系同源的方法被用于宏基因组reads的最后聚类。这个实验室还开发出了DiScRIBinATE (Ghosh et al., 2010) 【10】, ProViDE (Ghosh et al., 2011) 【11】, and SPHINX (Mohammed et al., 2011)【12】。
INDUS[14] and TWARIT[15]也是上述公司开发的,代表的序列组成的聚类方法,根据寡核苷酸组成来提高聚类的准确性。
一些其他的方法:
TACOA (Diaz et al., 2009)
Parallel-META (Su et al., 2011)
PhyloPythiaS (Patil et al., 2011)
RITA (MacDonald et al., 2012)[16]
MetaPhlAn (Segata et al., 2012) [17]
Treephyler
A tool for fast taxonomic profiling of metagenomes.
NBC
The Naïve Bayes Classification tool webserver for taxonomic classification of metagenomic...
CARMA
A software pipeline for characterizing the taxonomic composition and genetic diversity of...
MyTaxa
A homology-based bioinformatics framework to classify metagenomic and genomic sequences.
Sequedex
A signature-based method to classify the function and phylogeny of reads as short as 30 bp.
Genometa
A Java based local bioinformatics program which allows rapid analysis of metagenomic short read...
TaxSOM
A tool for taxonomic classification of DNA fragments, as they are typically obtained in...
Kraken (Taxonomic Sequence Classification System)
A system for assigning taxonomic labels to short DNA sequences, usually obtained through...
LMAT
Designed to efficiently assign taxonomic labels to as many reads as possible in very large...
RAIphy
A semi-supervised metagenomic fragment classification program.
ClaMS
A sequence composition-based classifier for metagenomic sequences.
MG-RAST
An automated analysis platform for metagenomes providing quantitative insights into microbial...
MARTA
This java-based software blasts each sequence that you provide it, and then looks for a consensus...
MLTreeMap
Analyzes DNA sequences and determines their most likely phylogenetic origin.
WebCARMA
Taxonomic classification of metagenomic shotgun sequences.
SOrt-ITEMS
Sequence orthology based approach for improved taxonomic estimation of metagenomic sequences.
Phymm and PhymmBL
Phylogenetic Classification of Metagenomic Data with Interpolated Markov Models.
ProViDE
A novel similarity based binning algorithm that uses a customized set of alignment parameter...
PhyloPythiaS
The Web Server for Taxonomic Assignment of Metagenome Sequences.
SPHINX
A hybrid binning approach that achieves high binning efficiency by utilizing both 'compositional'...
参考资料:
维基百科 http://en.wikipedia.org/wiki/Binning_(Metagenomics)
文献:Classification of metagenomic sequences: methods and challenges
