【2.3】序列拼接-velvet
一、安装
1.下载地址:http://www.ebi.ac.uk/~zerbino/velvet/
2.首先需要安装连个关联软件包zlib1g-dev 和pdflatex
sudo apt-get install zlib1g-dev (我的系统自带这个了)
sudo apt-get install texlive (这个得下)
3,解压下载的velvet软件包,
tar xvzf velvet_version.tgz
cd velvet_version
make color 'CATEGORIES=2' 'MAXKMERLENGTH=127' 'BIGASSEMBLY=1''LONGSEQUENCES=1' 'OPENMP=1'
(参数怎么修改见下面)
make clean 就是删除,然后再重新安装
其他一些设置:
make color 设置不同的颜色???
make 'MAXKMERLENGTH=127' #默认的最大kmer为31,这个需要修改
make 'CATEGORIES=57' #处理的数据的管道数,不同的管道可以用来处理来自不同插入文库或者不同样品(这里面有一个问题:宏基因组算是多个样品吗?),而这个值越大,对内存消耗越大,而我的样品,默认的为2,我怕内存不够,暂且设为2吧
make 'BIGASSEMBLY=1' #read的ID保存在一个32字节的字符串中,如果你有大于22亿(2.2G) 的reads,你就需要更多的内存,设置为1,是调用更多的内存。
make 'LONGSEQUENCES=1' #read的长度保存于16字节的字符串中,如果长度大于32kb,就意味着要更多的字节来存放字长度,所以可以直接设置为1,调用更多的内存,但是跟上面一样也会消耗很多的内存。
make 'OPENMP=1' #可以利用多个cpu,但是速度不会有指数的变化,因为只有一部分可以利用多线程
make ‘BUNDLEDZLIB=1’ #如果不能使用系统默认的zlib,也可以造常运行。
也可以直接这样,一致性到位: make color 'CATEGORIES=57' 'MAXKMERLENGTH=127' 'BIGASSEMBLY=1' 'LONGSEQUENCES=1' 'OPENMP=1'
环境变量:
export PATH=$PATH:/sam/velvet/bin
#注意我把安装好后的velveth,velvetg两个程序罗到了新建文件夹/bin里面,这样调用这两个程序的时候,就直接可以在终端的任何地方输入velveth,而不用cd 到程序目录,然后./velveth罗。
也可以这样来改环境变量:
sudo cp velvetg /usr/bin/
sudo cp velveth /usr/bin/
运行环境:
64bit,gcc(GNU Compiler Collection,GNU编译器套装,是自由的类Unix及苹果计算机Mac OS X 操作系统的标准编译器),>12G内存
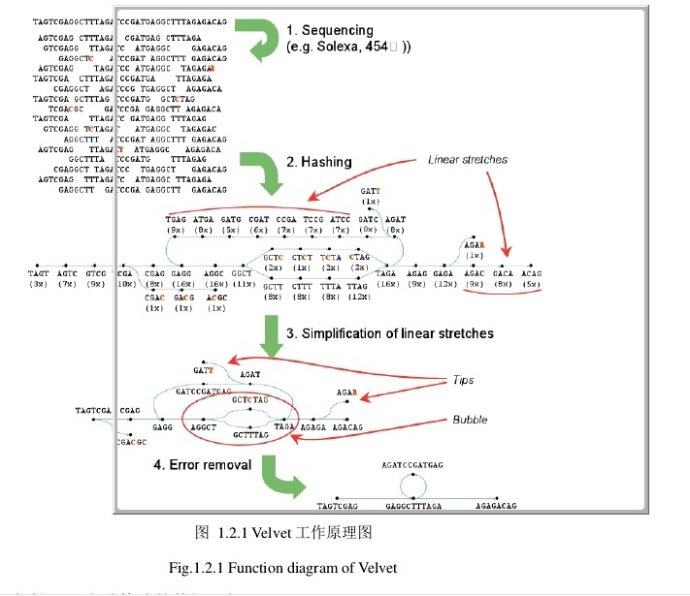
二、主要工作原理图:
1、Bruijn算法
给定的数据总是存在着冗余信息,因此,算法的使用很重要。当前大多数是用的overlap-consensus-layout算法,它的每一个read都是单独的实体。而Bruijn算法,将分析基于observed words(或者k-mers)。给定k-mers,不管发现多少次,都是唯一的node。此外,他可以无差别的容纳long和short read的混合物。 构建Bruijn图关键在于对所有的reads进行hash。Hash值通过设置参数给定,或者是默认的21。这对所有的序列进行成对比对时会减少时间,一旦所有的reads已经被hash,由k-mer node可以追溯他们的每一个路径。增值覆盖率和创建arcs就属于这种方法。
2、错误的移除
错误移除算法:Tour Bus,对没有错误连接的graph进行移除。这要确保基因组中低覆盖率的唯一的点没有被破坏。因此,这一步在画图之后进行。 这里有两种类型的错误:tips和bubbles。Tips:起源于低质量的read的结尾部分没有任何的重叠。Bubbles:他产生于长read的中间或两个错误的read结尾部分重叠。 首先去除tips,由长度和覆盖率来定义。然后去除bubbles,运用的是和Dijkstra’s算法相似的算法,最后低覆盖率的点会被淘汰。
三、使用
合并两个fastq文件,如果是fasta,则需将下面的代码中fastq改为fasta
/sam/velvet/contrib/shuffleSequences_fasta/shuffleSequences_fastq.pl output_forward_paired.fq output_reverse_paired.fq paired.fq
第一步:建立哈希索引
直接对fastq格式的原始文件进行处理,首先是用velveth命令建立hash表子集 输入./velveth会出来使用帮助:
./velveth directory hash_length {[-file_format][-read_type][-separate|-interleaved] filename1 [filename2 ...]} {...} [options]
directory : directory name for output files
hash_length : EITHER an odd integer (奇数)(if even, it will be decremented) <= 127 (if above, will be reduced)
: OR: m,M,s where m and M are odd(奇数) integers (if not, they will be decremented) with m < M <= 127 (if above, will be reduced)
and s is a step (even number). Velvet will then hash from k=m to k=M with a step of s
filename : path to sequence file or - for standard input
File format options:
-fasta -fastq -raw -fasta.gz -fastq.gz -raw.gz -sam -bam -fmtAuto
(Note: -fmtAuto will detect fasta or fastq, and will try the following programs for decompression : gunzip, pbunzip2, bunzip2(默认的是fasta格式)
File layout options for paired reads (only for fasta and fastq formats):
-interleaved : File contains paired reads interleaved in the one file (default)
-separate : Read 2 separate files for paired reads
Read type options:
-short
-shortPaired
-short2 独立的插入大小的文库
-shortPaired2 同上
-long -longPaired 针对sanger和454
-reference
Options:
-strand_specific : for strand specific transcriptome sequencing data (default: off)
原来这个可以针对转录组数据啊
-reuse_Sequences : reuse Sequences file (or link) already in directory (no need to provide original filenames in this case (default: off)
-reuse_binary : reuse binary sequences file (or link) already in directory (no need to provide original filenames in this case (default: off)
-noHash : simply prepare Sequences file, do not hash reads or prepare Roadmaps file (default: off)
-create_binary : create binary CnyUnifiedSeq file (default: off)
Synopsis:(大纲)
- Short single end reads:
velveth Assem 29 -short -fastq s_1_sequence.txt
- Paired-end short reads (PE1和PE2是合并的remember to interleave paired reads):
velveth Assem 31 -shortPaired -fasta interleaved.fna
# Assem为生成文件所在文件夹,31为设置的kmer,一般为奇数,默认的是31,最开始默认最大的也为31,我已经改为127了,-shortPaired双末端模式,interleaved.fna?难道是是它指定的排列顺序?默认的输入文件是fasta格式
- Paired-end short reads (PE1和PE2是分开的)
velveth Assem 31 -shortPaired -fasta -separate left.fa right.fa
- Two channels and some long reads:
velveth Assem 43 -short -fastq unmapped.fna -longPaired -fasta SangerReads.fasta
- Three channels:
velveth Assem 35 -shortPaired -fasta pe_lib1.fasta -shortPaired2 pe_lib2.fasta -short3 se_lib1.fa
Output:
directory/Roadmaps
directory/Sequences
[Both files are picked up by graph, so please leave them there]
第二步:根据建立的索引来组装
在终端输入./velvetg 查看说明
Usage:
./velvetg directory [options]
directory : working directory name
Standard options:
-cov_cutoff : removal of low coverage nodes AFTER tour bus or allow the system to infer it
(default: no removal)
-ins_length : expected distance between two paired end reads (default: no read pairing)
-read_trkg : tracking of short read positions in assembly (default: no tracking)
追踪段read的位置(默认的是关)
-min_contig_lgth : minimum contig length exported to contigs.fa file (default: hash length * 2)
-amos_file : export assembly to AMOS file (default: no export)
-exp_cov : expected coverage of unique regions or allow the system to infer it
(default: no long or paired-end read resolution)
-long_cov_cutoff : removal of nodes with low long-read coverage AFTER tour bus
(default: no removal)
Advanced options:
-ins_length* : expected distance between two paired-end reads in the respective short-read dataset (default: no read pairing)
-ins_length_long : expected distance between two long paired-end reads (default: no read pairing)
-ins_length*_sd : est. standard deviation of respective dataset (default: 10% of corresponding length)
[replace '*' by nothing, '2' or '_long' as necessary]
-scaffolding : scaffolding of contigs used paired end information (default: on)
-max_branch_length : maximum length in base pair of bubble (default: 100)
-max_divergence : maximum divergence rate between two branches in a bubble (default: 0.2)
-max_gap_count : maximum number of gaps allowed in the alignment of the two branches of a bubble (default: 3)
-min_pair_count : minimum number of paired end connections to justify the scaffolding of two long contigs (default: 5)
-max_coverage : removal of high coverage nodes AFTER tour bus (default: no removal)
-coverage_mask : minimum coverage required for confident regions of contigs (default: 1)
-long_mult_cutoff : minimum number of long reads required to merge contigs (default: 2)
-unused_reads : export unused reads in UnusedReads.fa file (default: no)
-alignments : export a summary of contig alignment to the reference sequences (default: no)
-exportFiltered : export the long nodes which were eliminated by the coverage filters (default: no)
-clean : remove all the intermediary files which are useless for recalculation (default : no)
-very_clean : remove all the intermediary files (no recalculation possible) (default: no)
-paired_exp_fraction : remove all the paired end connections which less than the specified fraction of the expected count (default: 0.1)
-shortMatePaired* : for mate-pair libraries, indicate that the library might be contaminated with paired-end reads (default no)
-conserveLong : preserve sequences with long reads in them (default no)
Output:
directory/contigs.fa : fasta file of contigs longer than twice hash length
directory/stats.txt : stats file (tab-spaced) useful for determining appropriate coverage cutoff
directory/LastGraph : special formatted file with all the information on the final graph
directory/velvet_asm.afg : (if requested) AMOS compatible assembly file
velvetg Assem -cov_cutoff 30 -ins_length 350 -ins_length_sd 100 -exp_cov auto -min_contig_lgth 500
#-ins_length为Insert size加上reads的长度,我前者为160,后者为91+91,所以后面的参数为350 ;默认的
#-ins_length_sd 100为什么设置这两个参数就有点不明白了,Assem注意和第一步建立的文件夹的对应
四、讨论
1.velvet拼接过程中主要的几个需要调试的参数
主要有三个:Kmer值,exp_cov和cov_cutoff三个。本文分别设置不同的参数值,进行比较。 Kmer值是基于De Bruijn算法的高通量读段拼接软件中最重要的一个参数,Kmer必须为奇数,在执行velveth时设置。在velvet的使用说明中,有一段关于Kmer设置的一般要求,如下: Ck= C(L-K+1)/L L表示读段长度,K表示Kmer值,C表示碱基的覆盖深度,Ck表示kmer的覆盖深度。L值已知,C值通过基因组规模和测序量来估计,当K值设定之后,可算得Ck值。根据经验,Ck值应当大于10,才能较好地完成拼接,而当Ck值大于20时,就浪费了测序深度。Kmer值越大Ck值就越小。当测序深度加大的时候,Kmer值也可以设的更大,有利于拼接。这个公式只能确定Kmer的取值范围,在此范围内选择采用哪个Kmer值,则需要进行Kmer参数值的调试。
在执行velvetg时,可设定exp_cov和cov_cutoff两个选项。选项cov_cutoff用来过滤低覆盖深度的contigs,选项exp_cov为预期的覆盖深度。这两个参数可设定具体的值,默认值为“auto”。当设为auto时,软件在拼接时自动取值,如果数据的覆盖比较均匀,设为auto,拼接结果会比较好;exp_cov设为auto时,cov_cutoff也将强制为auto,auto表示覆盖深度的阈值为exp_cov的一半。
2.如何使用多个Kmer值进行拼接
If you wish for Velvet to e?ciently test various k-mer lengths without doing redundant computations, you can request all k-mer lengths such that m ≤ k < M with a step of s:
> ./velveth output_directory/ m,M,s (..data files..)
几个小贴士:
1, velvet容许通道|,*代替任意字符等快捷方式
2, 可以将第一步分成两步,据说可以节约时间,不理解
> ./velveth output_directory/ 21 (..data files..) -noHash
> ./velveth output_directory/ 21 -reuse_Sequences
3,对于数据量很大,可以压缩包来读取
> ./velveth output_directory/ 21 -create_binary (..data files..)
##Ps:
修改MAXKMERLENGTH修改了半天,我在查看其MAXKMERLENGTH值的时候是cd到我的安装文件,然后velvet,这样能看到他的值,实际上这个值是环境变量的值,而不是我这个安装程序的值,我不知道我系统之前安装过嘛,最后只有sudo apt-get remove –auto-remove velvet,删除这个velet后,在重新安装,重新修改环境变量。通过这个折腾,我深刻了理解了./velveth和velveth这两个调用程序还是有本质的区别的. 最后感谢 QQ好友:生物信息学-小徐 不厌其烦的帮助我找问题,谢谢罗
参考资料:
阿飞与安达博客:http://liuwei441005.blog.163.com/blog/static/135705811201211751914615/
xiaoshijun的个人博客 http://blog.sciencenet.cn/blog-303373-715739.html
微斯人 http://mingkang1217.blog.163.com/blog/static/20352272011620250132/
百度文库 http://wenku.baidu.com/view/2884921dfc4ffe473368abf4.html
软件自带说明书 强烈推荐这个说明书,逻辑和易读性很强
