【1.2.4】人参考基因组版本(hg38/CRCH38..)
hg19,GRCH37和ensembl75是三种国际生物信息学数据库资源收集存储单位,即NCBI,UCSC和ENSEMBL各自发布的基因组信息。
下载链接: https://sapac.support.illumina.com/sequencing/sequencing_software/igenome.html
以下是不同机构之间参考基因组的对应关系:
| NCBI | UCSC | ENSEMBL |
|---|---|---|
| GRCh36 | hg18 | release_52 |
| GRCh37 | hg19 | ENSEMBL 59/61/64/68/69/75 |
| GRCh38 | hg38 | ENSEMBL 76/77/78/80/81/82 |
每次基因组版本的升级,比如从hg18到hg19,再到hg38,坐标系统已经不一样,所以分析过程中使用了某个基因组,去公共数据库查询频率,位置等信息时,都要对应到使用的参考基因组查询相关信息,才能保持信息的一致性。另外,如果需要,基因组坐标间也能通过LiftOver进行转换。
一、提供参考基因组的机构
The Genome Reference Consortium (GRC)
NCBI 旗下的 GRC 致力于为我们提供最完善的人类参考基因组。GRC 版本的基因组是 one-based coordinate system。除了基础的组装外,它还提供了 alternate loci 以表示那些过于复杂而无法用单条通路表示的区域。此外,它还以 patch 的形式修正某些区域,例如你会看到 GRCh37.p7 这样的版本号。通过这种方式,既可以保证整个染色体坐标的稳定性,同时提供更加准确的碱基组成。
UCSC
UCSC 本身并不会进行基因组的测序及组装,它只是将 GRC 的版本进行了一定的调整,例如在染色体编号之前加上 chr 等。需要注意的是 UCSC 给出的基因组版本是 zero-based coordinate system。 其他具体的调整我们会在接下来各版本参考基因组中进行更详细的说明。
ENSEMBL
ENSEMBLE 同样是使用来自 GRC 给出的参考基因组。与 UCSC 不同的是,其使用的是 one-based coordinate system。
二、基因组介绍
2.1 GRCh37/hg19/b37/ENSEMBL 59/61/64/68/69/75
GRCh37 中包含
- 24条基本完整的染色体序列,即 1-22, X, Y
- 完整的线粒体序列
- unlocalized sequences, 指知道具体来自哪条染色体,但具体坐标未知的序列
- unplaced sequences, 指无法确定来自于那条染色体的序列
- alternate loci, 这些序列包含人类基因组特定区域的 alternate representations 由于GRC给出这些序列时并没有给出标准的命名,也没有指定顺序,导致其他不同的组织采用各自的方式进行标记
hg19:
- 针对24条基本完整的染色体序列,使用 chr1-chr22, chrX and chrY 的命名
- 没有使用来自GRCh37的线粒体 NC_012920, 而是使用了老版本的 NC_001807,命名为 chrM
- 针对 unlocalized sequences, unplaced sequences, alternate loci 序列,给出了自定义的名称
- 用小写字母标记 repeats and low complexity regions, 即进行 soft-masked 尽管hg19版本存在不少的问题,但通过 UCSC genome browser 带来的大量曝光,hg19事实上应用广泛,包括不少外显子捕获试剂制造商利用该版本进行坐标定位
b37 (1000 Genomes Project Phase I):
- 24条基本完整的染色体序列,命名为 1-22, X, Y
- 使用来自GRCh37的线粒体,命名为 MT
- unlocalized sequences及unplaced sequences使用 accession number 命名,未包含 alternate loci 序列
- 这些约定在之后的 ENSEMBL genome browser, the NCBI dbSNP (in VCF files), the Sanger COSMIC (in VCF files) 等中得以继承。后续多数新的项目更倾向于该标准
b37+decoy / hs37d5 (1000 Genomes Project Phase II)
- 可以理解为b37的升级版,做了一些调整:
- A human herpesvirus (疱疹病毒) 4 type 1 sequence,命名为 NC_007605
- “decoy” sequence (诱饵序列)(命名为 hs37d5) 来自HuRef、BAC和质粒克隆和NA12878,可以提高序列比对的正确率
- Y染色体上 pseudo-autosomal regions (PAR) 被masked (用 N代替),所以X染色体上对应区域可能被视为二倍体
- 以上这些变化使得该套序列能够减少假阳性,且与b37兼容,从而成为序列比对和变异识别的最佳之选
2.2 GRCh38/hg38/ENSEMBL 76/77/78/80/81/82
相较于 GRCh37,GRCh38 的碱基组成以及坐标位置都有所变化,此外添加了更多的 ALT contig,因此两者之间存在不小的差异。从2013年12月以来,38版本的基因组 及相关的注释其实都已经很完善了,因此会建议新的项目都是用该版本的参考基因
2.3 人基因组fasta注释文件可以分为以下几部分序列
Primary assembly,包含以下三部分:
-
Assembled chromosomes:chr1-chr22,chrX,chrY和chrM的序列.
-
Unlocalized sequence:以_random结尾的序列,表示知道在哪条染色体上,但不知道方向和顺序.

- Unplaced sequence:以chrU_为前缀的序列,不知道在哪个染色体上.
Alternate contigs, alternate scaffolds或 alternate loci
以alt结尾的序列.用来表征单倍体序列的多样性,这是由于基因组是用单倍体类型表现的,比如1号染色体有两条,但fasta文件里只有一条的序列,由于基因的多样性(如等位基因)无法通过一条序列表示,所以就有了alt序列来补充说明. 但这样的alt序列在测序分析map的的过程中容易产生multiple-mapping低质量的 reads.而GATK的ZeroMappingQuality 会将这样的reads过滤掉.
PAR 区域
伪染色体序列(pseudoautosomal region),PAR区域的基因在X和Y染色体上都存在.但在map序列时会造成multiple-mapping reads,所以需要其中一条染色体(如y染色体)上的PAR区域mask掉.

decoy基因组
包含人疱疹病毒(EBV)基因组的序列.
2.4 关于基因组版本
在下载基因组文件的时候,可以发现即使是GRCh38版本,也有:GRCh38.p6,GRCh38.p11等小版本.这里的p是Patchs指定期对基因组的修补,并且每次修补并没有扰乱染色体位置信息.有两种patch:
- Fix patches是表示下次主版本发布时将要替换的序列.
- Novel patches表示上面提到的alternate loci.也就是将新的patches看做变异序列.
2.5 关于analysis set
在下载基因组文件时,常会看到analysis set的基因组文件:
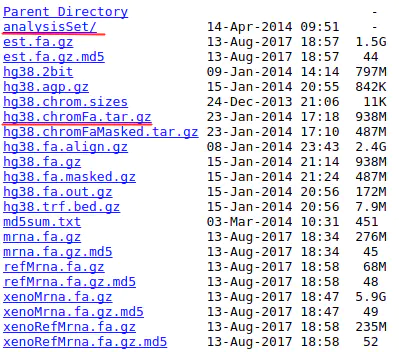
常用基因组文件只包含上面提到的Primary assembly,而analysis set还包含alt序列,PAR序列,decoy基因组.这些对于做基因组变异分析是必须的.可以看出笔者上面用来示范的那个GRCh38文件是analysis set.
三、下载
看起来NCBI也是很简单,就GRCh36,37,38,但是里面水也很深!
Feb 13 2014 00:00 Directory April_14_2003
Apr 06 2006 00:00 Directory BUILD.33
Apr 06 2006 00:00 Directory BUILD.34.1
Apr 06 2006 00:00 Directory BUILD.34.2
Apr 06 2006 00:00 Directory BUILD.34.3
Apr 06 2006 00:00 Directory BUILD.35.1
Aug 03 2009 00:00 Directory BUILD.36.1
Aug 03 2009 00:00 Directory BUILD.36.2
Sep 04 2012 00:00 Directory BUILD.36.3
Jun 30 2011 00:00 Directory BUILD.37.1
Sep 07 2011 00:00 Directory BUILD.37.2
Dec 12 2012 00:00 Directory BUILD.37.3
从上面可以看到,有37.1, 37.2和 37.3 等等,不过这种版本一般指的是注释在更新而基因组序列一般不变。
总之你需要记住, hg19基因组大小是3G,压缩后八九百兆。
如果要下载GTF注释文件,基因组版本尤为重要。
NCBI:最新版(hg38)
NCBI:其它版本
Ensembl
变化上面链接中的release就可以拿到所有版本信息
UCSC
它本身需要一系列参数:
1. Navigate to http://genome.ucsc.edu/cgi-bin/hgTables
2. Select the following options:
clade: Mammal
genome: Human
assembly: Feb. 2009 (GRCh37/hg19)
group: Genes and Gene Predictions
track: UCSC Genes
table: knownGene
region: Select "genome" for the entire genome.
output format: GTF - gene transfer format
output file: enter a file name to save your results to a file, or leave blank to display results in the browser
3. Click 'get output'.
搞清楚版本关系了,接下来就是进行下载。UCSC里面下载非常方便,只需要根据基因组简称来拼接url:
http://hgdownload.cse.ucsc.edu/goldenPath/mm10/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/mm9/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/chromFa.tar.gz
或者用shell脚本指定下载的染色体号
for i in $(seq 1 22) X Y M;
do echo $i;
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr${i}.fa.gz;done
gunzip *.gz
for i in $(seq 1 22) X Y M;
do cat chr${i}.fa >> hg19.fasta;
done
rm -fr chr*.fasta
四、讨论
4.1 decoy的意义
参考基因组是不完整的,特别是在着丝粒周围,因此,真正属于其他地方的读段常常被错误地映射到基因组中的特定位置,因为参考中缺少真正的匹配。 这些会引起误报,这在“ 1000基因组计划”中给我们带来困扰。 诱饵(decoy)是对此的一种务实解决方案-它包含已知的真实人类基因组序列,该序列不在参考基因组中,并且会“吸取”读数,否则将以低质量映射到参考物中。 诱饵是由Broad的Heng Li与Richard Durbin(桑格)和Genome Reference Consortium(维护参考基因组)的Deanna Church合作建造的。
4.2 Which one to choose?
BWA 作者 Heng Li 在其博客中给出了他的建议:
-
如果使用37版本的参考基因组,则选择 hs37-1kg: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/human_g1k_v37.fasta.gz
-
如果使用37版本的参考基因组,且认为诱饵序列有助于之后的分析,则选择 hs37d5: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz
-
如果使用的是38版本的参考基因组,则选择: ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz
他列举了不同版本参考基因组中存在的问题,包括:
-
包含 ALT contig。这些序列中有很大部分侧翼序列与 primary human assembly 中的序列基本一致,大部分序列比对软件给到这些区域的 map quality 值为0,这就导致了后续变异识别以及其他分析的敏感性下降。可以通过选择 ALT-aware 的软件解决这个问题,但目前主流的软件并不支持
-
Padding ALT contigs,当中有大量的 N 碱基,类似于1带来的问题 (用很长的N间隔这些ALT contig序列,增加了不必要的ref的size)
-
包含 multi-placed 的序列。例如 X 染色体上的 PAR 也放在 Y 染色体上。如果使用同时包含这两部分的参考基因组,就无法使用标准流程对这块区域进行变异识别。还有像在 GRCh38 中,部分 alpha satellites 被放置多次。正确的解决方法时间 Y 染色体上的 PAR 区域以及 alpha repeats 多出来的拷贝 hard maseked
-
rCRS 线粒体序列的使用。rCRS 在群体遗传性中大量使用,而 GRCh37 所使用的线粒体序列要比它长2bp,这可能导致在进行线粒体系统进化分析时出现问题。GRCh38 使用的则是 rCRS
-
将半模糊的 IUB codes 转换成了 N,虽然总体而言影响不大,因为人类染色体序列中只有极少部分这样的碱基
-
用 accession number 代替染色体名字进行命名,不够直观
-
没有包含 unplaced and unlocalized contigs
具体到个版本参考基因组,问题就是这样的:
- hg19/chromFa.tar.gz from UCSC: 1, 3, 4 and 5
- hg38/hg38.fa.gz from UCSC: 1, 3 and 5
- GCA_000001405.15_GRCh38_genomic.fna.gz from NCBI: 1, 3, 5 and 6
- Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz from EnsEMBL: 3
- Homo_sapiens.GRCh38.dna.toplevel.fa.gz from EnsEMBL: 1, 2 and 3
当然,怎么选择参考基因组还是需要根据具体的情况来判定,包括实验室或者公司之前使用的情况,不同版本对应的注释文件情况等等
参考资料
- http://www.biotrainee.com/thread-411-1-1.html
- https://chendianyu.github.io/ngs/2018/05/29/human-reference-genome-and-annotation/
- https://www.biostars.org/p/73100/
- https://lh3.github.io/2017/11/13/which-human-reference-genome-to-use
- https://gatk.broadinstitute.org/hc/en-us/articles/360035890951-Human-genome-reference-builds-GRCh38-or-hg38-b37-hg19
