生信分析软件包--emboss
一、简介
EMBOSS是“欧洲分子生物学开放软件套件”。 EMBOSS是一个免费的开源软件分析软件包,专为分子生物学(例如EMBnet)用户社区的需求而开发。 该软件自动处理各种格式的数据,甚至允许从网络透明检索序列数据。 此外,随着软件包提供了广泛的库,它是一个允许其他科学家以真正的开源精神开发和发布软件的平台。 EMBOSS还将一系列当前可用的包和工具集成到一个无缝整体中进行序列分析。 EMBOSS打破了商业软件包的历史趋势
1.1 特点
- 一个正确构建的工具包,用于创建强大的生物信息学应用程序或工作流程。
- 一套全面的序列分析程序。
- 处理所有序列和许多对齐和结构格式。
- 用于常见序列分析任务的丰富编程库。
- 许多其他领域的附加编程库,包括字符串处理,模式匹配,列表处理和数据库索引。
- 这个是免费的。
- 这是一个开源项目。
- 它几乎可以运行在你能想到的每一个UNIX上,有些是你不能运行的,还有MS Windows和MacOS。
- 每个应用程序都具有相同的界面风格,因此掌握了一个,您已经掌握了所有这些。
- 一致的用户界面使GUI设计人员和开发人员更加便利。
- 它集成了其他流行的公开包。
- 它没有任意大小限制:可以处理的数据量没有限制。 对于程序员来说,简化了序列和数组等对象的内存管理。
2.1 可以用来干什么
- 序列比对,
- 使用序列模式快速搜索数据库,
- 蛋白质基序识别,包括域分析,
- 核苷酸序列模式分析 - 例如识别CpG岛或重复,
- 小基因组的密码子使用分析,
- 快速识别大规模序列集中的序列模式,
- 出版的演示工具
常用的个工具(可是远远远远不止这些工具呀。。)
prophet Gapped alignment for profiles.
infoseq Displays some simple information about sequences.
water Smith-Waterman local alignment.
pepstats Protein statistics.
showfeat Show features of a sequence.
palindrome Looks for inverted repeats in a nucleotide sequence.
eprimer3 Picks PCR primers and hybridization oligos.
profit Scan a sequence or database with a matrix or profile.
extractseq Extract regions from a sequence.
marscan Finds MAR/SAR sites in nucleic sequences.
tfscan Scans DNA sequences for transcription factors.
patmatmotifs Compares a protein sequence to the PROSITE motif database.
showdb Displays information on the currently available databases.
wossname 在一行文档中按关键字查找程序
abiview Reads ABI file and display the trace.
tranalign Align nucleic coding regions given the aligned proteins.
二、安装
cd /data/src
wget ftp://ftp.ebi.ac.uk/pub/software/unix/EMBOSS/EMBOSS-6.6.0.tar.gz
gunzip EMBOSS-6.6.0.tar.gz
tar xvf EMBOSS-6.6.0.tar
cd EMBOSS-6.6.0
./configure -prefix=/data/software/EMBOSS-6.6.0
make -j12
三、使用说明
3.1 wossname 通过关键词来搜索相关的关键
在终端输入wossname,后即可输入关键词来搜索相关程序,如果不输入,则显示所有的程序(我的妈呀,这里面包含的程序真多)
[root@c04 bin]# wossname
Find programs by keywords in their short description
Text to search for, or blank to list all programs:
具体的例子,当输入“protein”以后,则有这么多软件
Finds programs by keywords in their one-line documentation
Keyword to search for: protein
SEARCH FOR 'PROTEIN'
antigenic Finds antigenic sites in proteins
backtranseq Back translate a protein sequence
checktrans Reports STOP codons and ORF statistics of a protein sequence
emowse Protein identification by mass spectrometry
digest Protein proteolytic enzyme or reagent cleavage digest
eprotdist Protein distance algorithm
eprotpars Protein parsimony algorithm
fuzzpro Protein pattern search
fuzztran Protein pattern search after translation
garnier GARNIER predicts protein secondary structure.
iep Calculates the isoelectric point of a protein
octanol Displays protein hydropathy
oddcomp Finds protein sequence regions with a biased composition
patmatdb Search a protein sequence database with a motif
patmatmotifs Search a motif database with a protein sequence
pepnet Displays proteins as a helical net
pepstats Protein statistics
pepwheel Shows protein sequences as helices
pepwindow Displays protein hydropathy
pepwindowall Displays protein hydropathy of a set of sequences
preg Regular expression search of a protein sequence
pscan Scans proteins using PRINTS
sigcleave Reports protein signal cleavage sites
topo Draws an image of a transmembrane protein
当然,你可以通过参数来限定你想要的程序
wossname -opt
现在,您将看到各种其他选项。 每个选项的默认值在方括号中给出,您可以按return键接受默认值,也可以输入所需的值
Keyword to search for: protein
Output program details to a file [stdout]: myfile
Format the output for HTML [N]: Y
String to form the first half of an HTML link:
String to form the second half on an HTML link:
Output only the group names [N]:
Output an alphabetic list of programs [N]:
Use the expanded group names [N]:
这组命令将使wossname将程序列表写入名为myfile的文件,格式为HTML格式,可以在Web浏览器中查看。
要生成所有当前EMBOSS程序的列表,请再次启动wossname,但不要指定关键字,请按return。 程序列表将滚动到您的屏幕上,根据其功能分组。 向上和向下滚动以查看所有内容。 你能想到如何将这些数据存入文件吗? (提示:使用-opt)
如果将标志-help附加到任何EMBOSS程序的名称,您将看到该程序可用的所有命令标志的列表。 例如:
wossname -help
3.2 序列分析
在本教程中,我们将研究视紫红质家族G蛋白偶联受体的成员。 当然,一般原则适用于您想要分析的任何序列。 我们将处理从EMBL和SwissProt检索的序列,但您也可以在文本文件中使用EMBOSS序列。
我们将从两个标识为XL23808和XLRHODOP的EMBL序列开始; 这些序列是非洲爪蟾视紫红质的基因组和相应的cDNA序列。
您需要告诉EMBOSS在哪里读取您想要分析的序列。 EMBOSS可以从文本文件或直接从序列数据库中读取序列。 最简单的方法是使用示例。
3.2.1 从数据库中检索序列
EMBOSS程序可以从各种序列数据库中读取序列,前提是在表格数据库中输入序列:条目。 这种格式称为USA(统一序列地址)。 有关美国的更多信息,请访问EMBOSS网站。 您可以使用showdb程序查看我们为您设置的数据库:
例如,以下是在HGMP中使用EMBOSS的前几个数据库。 根据当地EMBOSS维护者的设置,您的本地站点可能会有不同的数据库选择。
[root@c04 bin]# showdb
Display information on configured databases
# Name Type Comment
# ============= ======== =======
taxon Taxonomy NCBI taxonomy
drcat Resource Data Resource Catalogue
chebi Obo Chemical Entities of Biological Interest
eco Obo Evidence code ontology
edam Obo EMBRACE Data and Methods ontology
edam_data Obo EMBRACE Data and Methods ontology (data)
edam_format Obo EMBRACE Data and Methods ontology (formats)
edam_identifier Obo EMBRACE Data and Methods ontology (identifiers)
edam_operation Obo EMBRACE Data and Methods ontology (operations)
edam_topic Obo EMBRACE Data and Methods ontology (topics)
go Obo Gene Ontology
go_component Obo Gene Ontology (cellular components)
go_function Obo Gene Ontology (molecular functions)
go_process Obo Gene Ontology (biological processes)
pw Obo Pathways ontology
ro Obo Relations ontology
so Obo Sequence ontology
swo Obo Software ontology
3.2.2 seqret
seqret按顺序读取并写出来,实际上是EMBOSS等效于readseq。 它可能是最常用的EMBOSS程序
unix % seqret
Reads and writes (returns) a sequence
Input sequence: embl:xlrhodop
Output sequence [xlrhodop.fasta]:
unix % more xlrhodop.fasta
>XLRHODOP L07770 Xenopus laevis rhodopsin mRNA, complete cds.
ggtagaacagcttcagttgggatcacaggcttctagggatcctttgggcaaaaaagaaac
acagaaggcattctttctatacaagaaaggactttatagagctgctaccatgaacggaac
这个功能更多的是从数据库中提取序列,后续再来整理和理解。。
3.2.3 从文件中读取序列,并转换文件类型
seqret xlrhodop.fasta -outseq gcg::myseq.gcg
或
seqret xlrhodop.fasta -outseq myseq.gcg -osformat gcg
3.2.4 获得序列信息
infoseq embl:xlrhodop
Displays some simple information about sequences
# USA Name Accession Type Length GC Description
embl-id:XLRHODOP XLRHODOP L07770 N 1684 45.72 X.laevis rhodopsin
3.2.x 获得多序列
infoseq
Displays some simple information about sequences
Input sequence(s): sw:opsd_*
# USA Name Accession Type Length Description
sw-id:OPSD_ABYKO OPSD_ABYKO O42294 P 289 RHODOPSIN (FRAGMENT).
sw-id:OPSD_ALLMI OPSD_ALLMI P52202 P 352 RHODOPSIN.
sw-id:OPSD_AMBTI OPSD_AMBTI Q90245 P 354 RHODOPSIN.
sw-id:OPSD_ANGAN OPSD_ANGAN Q90214 P 352 RHODOPSIN, DEEP-SEA
sw-id:OPSD_ANOCA OPSD_ANOCA P41591 P 352 RHODOPSIN.
sw-id:OPSD_APIME OPSD_APIME Q17053 P 377 RHODOPSIN.
sw-id:OPSD_ASTFA OPSD_ASTFA P41590 P 352 RHODOPSIN.
sw-id:OPSD_BATMU OPSD_BATMU O42300 P 289 RHODOPSIN (FRAGMENT).
sw-id:OPSD_BATNI OPSD_BATNI O42301 P 289 RHODOPSIN (FRAGMENT).
sw-id:OPSD_BOVIN OPSD_BOVIN P02699 P 348 RHODOPSIN.
我们也可以在命令行中使用通配符,但是在这里我们必须用引号将规范括起来
infoseq ``sw:opsd_*''
您可以使用secret将多个序列检索到文件中; 例如:
seqret ``sw:opsd_a*'' -outseq opsd_a.seqs
检索标识符以“opsd_a”开头的所有序列到名为opsd_a.seqs的文件中。 如果我们想将每个序列放在一个单独的文件中,我们可以输入:
seqret ``sw:opsd_a*'' -ossingle
3.3 两条序列的比对
本章是关于序列相似性的。 让我们从警告开始:没有独特,精确或普遍适用的相似概念。 比对是两个序列的排列,其显示两个序列相似的位置,以及它们不同的位置。 当然,最佳对齐是表现出最显着的相似性和最小差异的对齐。 概括地说,序列比较有三类方法。
- Dotplots:片段方法将来自一个序列的所有窗口(预定长度的重叠片段(例如,10个氨基酸))与来自另一个序列的所有片段进行比较。 这是点图中使用的方法。
- Global alignment: 最佳的全局比对方法允许获得用于比较两个序列的最佳总分,包括考虑间隙。
- Local alignment: 最佳局部对齐算法寻求识别两个序列之间的最佳局部相似性,但与段方法不同,包括对间隙的明确考虑。
Dotplots
两个序列之间比较的最直观表示使用点图。 在每个轴上表示一个序列,并且沿着矩阵中的对角线分布显着的匹配区域。
dottup
DNA sequence dot plot
Input sequence: embl:xl23808
Second sequence: embl:xlrhodop
Word size [4]: 10
Graph type [x11]:
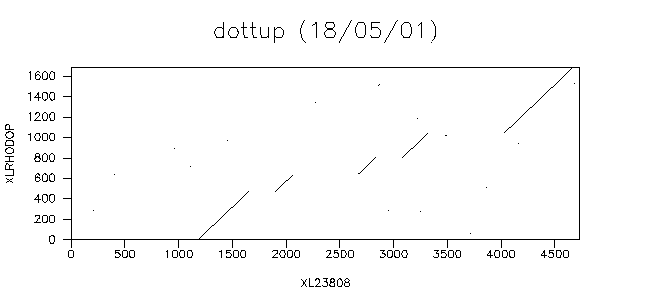
对角线表示两个序列良好对齐的区域。 你可以看到有五个清晰的对角线。 你会记得我们正在调整基因组和cDNA - 这五个对角线代表基因的五个外显子! 如果您使用SRS查看基因组序列的原始EMBL条目,您将看到带注释的条目表明该基因中有五个外显子。 所以我们的结果是一致的。
我们在此示例中使用的设置是那些可以获得最佳结果的设置。 dottup查找序列之间的完全匹配。 由于我们期望基因组序列中的外显子区域与cDNA序列完全匹配,我们可以使用更长的字长,因为我们仍然可以获得完全匹配。 这给出了一个非常干净的情节。 如果您要将cDNA序列与相关序列的cDNA序列匹配,例如 来自鼠标的视紫红质(embl:m55171)那么你不会期望长的完全匹配,所以应该使用较短的字长。
dottup embl:m55171 embl:xlrhodop
DNA sequence dot plot
Word size [4]: 10
Graph type [x11]:
- 对于使用不同字长的两种比较重复此操作。你注意到什么?
- 哪个字大小给出了最清晰plot?
- 为什么第一个和最后一个外显子中的对角线不那么清晰?
点图不会给我们任何详细的序列信息。为此,我们需要使用不同的程序。我们将使用的算法比用于搜索数据库的算法更严格;因此,即使您使用类似BLAST的数据库从数据库中检索了序列,在您之后进行仔细的成对比对时也是值得的。序列比对程序背后的基本思想是以这样的方式对齐两个序列以产生最高分数 - 评分矩阵用于为每个匹配的分数添加点并且针对每个不匹配减去它们。用于核酸比对的基质倾向于涉及相当简单的匹配/错配评分方案,而通常用于评分蛋白质比对的基质更复杂,其中评分旨在反映不同氨基酸之间的相似性而不是简单地评分同一性。随着时间的推移,各种突变发生在序评分矩阵试图应对突变,但插入和缺失需要一些额外的参数以允许在比对中引入缺口。对于创造差距和扩大现有差距都有惩罚;已经发现在对齐程序中给出的默认间隙参数对于测试序列在经验上是正确的,但是您应该尝试不同的间隙罚分。
Global alignment
全局比对是在整个长度上比较两个序列的比对,并且适合于比较预期在整个长度上共享相似性的序列。 对齐使用提供给程序的评分矩阵和间隙参数最大化相似区域并最小化间隙。 EMBOSS程序针是针对全局对齐的Needleman-Wunsch []算法的实现; 如果序列很长,计算是严格的,并且针可能是耗时的。
[root@c04 bin]# needle
Needleman-Wunsch global alignment.
Input sequence:embl:xlrhodop
Second sequence:embl:xl23808
Gap opening penalty [10.0]:
Gap extension penalty [0.5]:
Output file [xlrhodop.needle]:
[root@c04 bin]# more xlrhodop.needle
Global: XLRHODOP vs XL23808
Score: 7471.00
XLRHODOP
XL23808 1 cgtaactaggaccccaggtcgacacgacaccttccctttcccagt 45
XLRHODOP
XL23808 46 tatttcccctgtagacgttagaaggggaaggggtgtacttatgtc 90
XLRHODOP
XL23808 91 acgacgaactacgtccttgactacttagggccagagagacgaggt 135
$ \vdots$
请注意,由于这是一个全局比对,整个基因组序列在输出中给出,即使在它不与cDNA对齐的区域也是如此。 向下滚动输出,直到到达对齐区域。
XLRHODOP 1 ggtagaacagcttcagttgggatcacaggcttcta 35
||||||||||||||||||||||||||||||||||
XL23808 1171 tgggtcatactgtagaacagcttcagttgggatcacaggcttcta 1215
XLRHODOP 36 gggatcctttgggcaaaaaagaaacacagaaggcattctttctat 80
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1216 gggatcctttgggcaaaaaagaaacacagaaggcattctttctat 1260
XLRHODOP 81 acaagaaaggactttatagagctgctaccatgaacggaacagaag 125
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1261 acaagaaaggactttatagagctgctaccatgaacggaacagaag 1305
XLRHODOP 126 gtccaaatttttatgtccccatgtccaacaaaactggggtggtac 170
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1306 gtccaaatttttatgtccccatgtccaacaaaactggggtggtac 1350
$ \vdots$
我们只显示了部分输出,因为它很长。 您应该查看整个输出,并注意有五个对齐的区域代表从点图预测的五个外显子。 仔细查看对齐区域的边界。 我们知道,作为生物学家,内含子/外显子边界具有保守的gt..ag对二核苷酸,限定了剪接位点。 由于它不了解基因结构模型,因此针头最不可能正确对齐这些边界。 它使用的评分方法并不专门处理剪接网站。 程序est2genome具有额外的评分因子,可以更好地对齐内含子/外显子边界。
Local alignment
如上所述,全局序列比对算法在整个序列上对齐序列。 您需要考虑这种类型的对齐是否对您的序列有意义。 对于我们的例子,我们期望每个外显子在序列中以相同的顺序表示,它运作良好 - 但是,您认为这种方法的效果如何,例如,共享一个域的多域蛋白质,但不是 其他,或有重复区域的序列?
第二种比较方法,即局部比对,搜索局部相似性的区域,并且不需要包括序列的整个长度。 局部对齐方法对于扫描数据库非常有用,或者当您不知道序列在整个长度上相似时。 EMBOSS程序水是用于局部比对的Smith Waterman算法的严格实现[]。
[root@c04 bin] water
Smith-Waterman local alignment.
Input sequence: embl:xlrhodop
Second sequence: embl:xl23808
Gap opening penalty [10.0]:
Gap extension penalty [0.5]:
Output file [xlrhodop.water]:
[root@c04 bin]# more xlrhodop.water
Local: XLRHODOP vs XL23808
Score: 7448.00
XLRHODOP 2 gtagaacagcttcagttgggatcacaggcttctagggatcctttg 46
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1182 gtagaacagcttcagttgggatcacaggcttctagggatcctttg 1226
XLRHODOP 47 ggcaaaaaagaaacacagaaggcattctttctatacaagaaagga 91
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1227 ggcaaaaaagaaacacagaaggcattctttctatacaagaaagga 1271
XLRHODOP 92 ctttatagagctgctaccatgaacggaacagaaggtccaaatttt 136
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1272 ctttatagagctgctaccatgaacggaacagaaggtccaaatttt 1316
XLRHODOP 137 tatgtccccatgtccaacaaaactggggtggtacgaagcccattc 181
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1317 tatgtccccatgtccaacaaaactggggtggtacgaagcccattc 1361
XLRHODOP 182 gattaccctcagtattacttagcagagccatggcaatattcagca 226
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1362 gattaccctcagtattacttagcagagccatggcaatattcagca 1406
XLRHODOP 227 ctggctgcttacatgttcctgctcatcctgcttgggttaccaatc 271
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1407 ctggctgcttacatgttcctgctcatcctgcttgggttaccaatc 1451
XLRHODOP 272 aacttcatgaccttgtttgttaccatccagcacaagaaactcaga 316
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1452 aacttcatgaccttgtttgttaccatccagcacaagaaactcaga 1496
XLRHODOP 317 acacccctaaactacatcctgctgaacctggtatttgccaatcac 361
|||||||||||||||||||||||||||||||||||||||||||||
XL23808 1497 acacccctaaactacatcctgctgaacctggtatttgccaatcac 1541
$ \vdots$
EMBOSS包含其他成对比对程序 stretcher和matcher 分别是全局和局部对齐程序,不像needle and water那么严格,因此运行得更快; 它们可能对数据库搜索很有用。 supermatcher设计用于非常大的序列的局部比对,并且在其实现中甚至不那么严格。 所有这些程序的文档页面都可以在以下位置找到 http://emboss.sourceforge.net/apps/
3.4 蛋白分析
本章将向您介绍一些可用于分析蛋白质序列的EMBOSS应用程序。 显然,前一章中说明的具有核酸序列的成对序列比较方法也可以与蛋白质序列一起使用。
3.4.1 鉴定ORF区域
在本节中,我们将向您展示一些简单的EMBOSS应用程序,用于将cDNA序列翻译成蛋白质。 您应该意识到基因结构预测是一个棘手的问题,并且识别基因组序列中的外显子/内含子边界并不容易; 就目前而言,我们将在实际应用中使用cDNA序列,而不是处理预测方面的问题。 首先,我们需要确定我们的开放阅读框架。 我们可以使用EMBOSS程序plotorf快速直观地了解我们序列的六个框架中ORF的分布。
[root@c04 bin] plotorf
Plot potential open reading frames
Input sequence: embl:xlrhodop
Graph type [x11]:
您将看到一个图形输出,显示所有六个帧中的潜在开放阅读框(ORF):

最长的ORF位于第2帧,位于100到1200左右。我们现在将确定翻译的确切起点和终点。 为此,我们可以使用EMBOSS程序getorf。
[root@c04 bin] getorf -opt
Finds and extracts open reading frames (ORFs)
Input sequence: embl:xlrhodop
Output sequence [xlrhodop.orf]:
Genetic codes
0 : Standard
1 : Standard (with alternative initiation codons)
2 : Vertebrate Mitochondrial
3 : Yeast Mitochondrial
4 : Mold, Protozoan, Coelenterate Mitochondrial and Mycoplasma/Spiroplasma
5 : Invertebrate Mitochondrial
6 : Ciliate Macronuclear and Dasycladacean
9 : Echinoderm Mitochondrial
10 : Euplotid Nuclear
11 : Bacterial
12 : Alternative Yeast Nuclear
13 : Ascidian Mitochondrial
14 : Flatworm Mitochondrial
15 : Blepharisma Macronuclear
Code to use [0]:
Minimum nucleotide size of ORF to report [30]:
Type of sequence to output
0 : Translation of regions between STOP codons
1 : Translation of regions between START and STOP codons
2 : Nucleic sequences between STOP codons
3 : Nucleic sequences between START and STOP codons
4 : Nucleotides flanking START codons
5 : Nucleotides flanking initial STOP codons
6 : Nucleotides flanking ending STOP codons
Type of output [0]: 3
请注意,您可以指定其密码子使用表最适合您序列的生物体,您还可以选择报告给您的信息类型。 在我们的例子中,我们只关心这个序列的起始密码子和终止密码子的位置。
plotorf只是getorf生成的文本信息的图形表示。 由于我们要求报告所有高于最小大小的ORF,getorf告诉我们许多潜在的ORFs。 我们从plotorf知道我们的ORF将在100到1200区域内,所以滚动输出文件xlrhodop.orf,直到你发现它为止。 实际的开始和结束位置是什么?
3.4.2 翻译序列
从上一次练习中你应该发现我们的cDNA序列中要翻译的区域是110到1171。 现在我们可以使用transeq翻译该区域并使用翻译的肽进行进一步分析。
让我们再次练习使用命令行标志(限定符)。 这里新的是-sbegin和-send。 这些允许您指定序列的子区域; 在这种情况下,我们将要求transeq仅转换我们已识别为编码区域的embl:xlrhodop部分。 你应该记得第2章中的-outseq。
[root@c04 bin] transeq embl:xlrhodop -sbegin 110 -send 1171 -outseq xlrhodop.pep
Translate nucleic acid sequences
[root@c04 bin] more xlrhodop.pep
>XLRHODOP+1 Xenopus laevis rhodopsin mRNA, complete cds.
MNGTEGPNFYVPMSNKTGVVRSPFDYPQYYLAEPWQYSALAAYMFLLILLGLPINFMTLF
VTIQHKKLRTPLNYILLNLVFANHFMVLCGFTVTMYTSMHGYFIFGQTGCYIEGFFATLG
GEVALWSLVVLAVERYMVVCKPMANFRFGENHAIMGVAFTWIMALSCAAPPLFGWSRYIP
EGMQCSCGVDYYTLKPEVNNESFVIYMFIVHFTIPLIVIFFCYGRLLCTVKEAAAQQQES
ATTQKAEKEVTRMVVIMVVFFLICWVPYAYVAFYIFTHQGSNFGPVFMTVPAFFAKSSAI
YNPVIYIVLNKQFRNCLITTLCCGKNPFGDEDGSSAATSKTEASSVSSSQVSPA
我们之前看到,这种蛋白质的SwissProt条目具有标识符opsd_xenla; 到目前为止测试您对EMBOSS的理解,使用针将您翻译的产品与数据库序列进行比较。 使用SRS将您的发现与SwissProt条目进行比较。
/nfs/public/ro/es/appbin/linux-x86_64/EMBOSS-6.6.0/bin/transeq emboss_transeq-I20210617-093439-0314-20317044-p1m.sequence -auto -stdout -frame 1 -table 0
- frame 6 表示6重翻译的结果都显示出来。
- -auto 表示直接出结果,省略一步步确认的交互功能
- -stdout 显示在屏幕上
- -table 输出的格式
具体的参数说明,可以参阅:
/nfs/public/ro/es/appbin/linux-x86_64/EMBOSS-6.6.0/bin/transeq --help
3.4.3 USA for partial sequences
作为-sbegin和-send的替代,您可以指定start,end以及是否将补语作为序列USA的一部分进行反转。 要使用的格式是db:sequence [start:end](或db:sequence [start:end:r]以反向补码)。 开始必须小于结束。 如果要使用实际的开始和结束,则使用值0而不是位置。 如果您想从序列的末尾而不是从头开始计数,则使用负数。
Residues 10-20 sw:opsd_xenla[10:20]
The last ten residues sw:opsd_xenla[-10:0]
The last twenty residues bar 5 sw:opsd_xenla[-20:-6]
bases 134-458 reverse complement embl:xlrhodop[134:458:r]
3.4.4 二级结构预测
自从确定问题以来,DNA序列如何决定特定蛋白质结构的问题一直是迷恋和猜测的源泉。 它仍然是一个非常困难的领域; 通常被称为“折叠问题”,它是分子生物学中的一个主要突出问题。 已经进行了许多尝试以从其序列预测蛋白质的三级结构。 这些分为两种截然不同的方法:
- 一种方法是建立蛋白质链的现实机械模型并模拟折叠过程。 *其他方法是经验性的,因为它们通过已知三级结构的推断来进行。
基于机械模型的结构预测方法具有先天(可能是致命的)吸引力,理论上,它不需要蛋白质三级结构的先验知识。 如果成功,它可以统一应用于所有序列。 相比之下,基于来自已知结构的推断的所有方法在其适用性方面具有固有的限制。 它们仅适用于预测与推理过程中使用的结构类似的结构。 幸运的是,通常有生物物理或生物化学线索有助于做出这一决定,这些线索通常被整合到结构预测方法中。
目前,实现合理二级结构预测的最佳方法是在序列上运行各种预测算法,并确定结果之间的一致性。 有各种Web服务器可以为您进行这些多重分析,包括HGMP的PIX和Dundee大学的Jpred:
pepinfo
pepinfo产生有关氨基酸特性(大小,极性,芳香性,电荷等)的信息。 疏水性谱也是可用的,可用于定位转角,潜在的抗原肽和跨膜螺旋。 采用各种算法,包括Kyte和Doolittle亲水测量 - 该曲线是在9个残基的窗口上的残基特异性疏水性指数的平均值。 当线在框架的上半部分时,它表示疏水区域,而当它在下半部分时,表示亲水区域。
[root@c04 bin] pepinfo xlrhodop.pep
Plots simple amino acid properties in parallel
Graph type [x11]:
Output file [pepinfo.out]:
You will see two screens (press return to move from the first to the second screen) that look like this:


3.4.5 预测跨膜区域
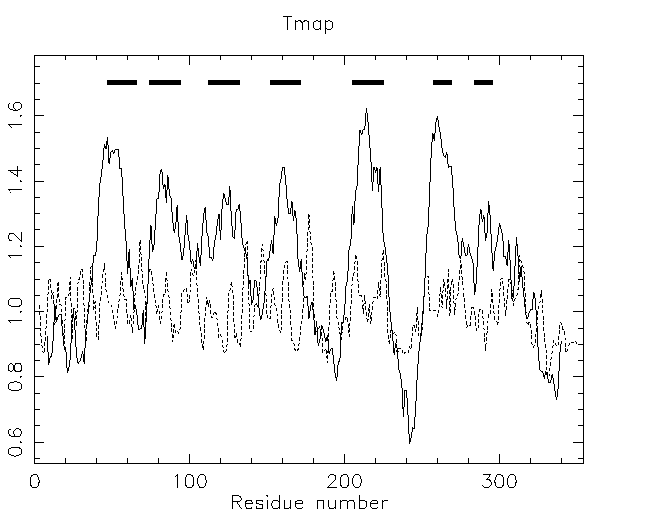
pepinfo亲水性图的结果显示xlrhodop.pep内有7个高度疏水的区域。 这些可能是跨膜结构域吗? 我们可以使用EMBOSS程序tmap来研究这种可能性:
[root@c04 bin] tmap
Displays membrane spanning regions
Sequences file to be read in: xlrhodop.pep
Graph type [x11]:
参考资料
