【2.1.2】reads mapping and variants calling
Reads mapping是指将测序得到的DNA片段(也就是reads)定位在基因组上,通过Reads mapping,在克服了深度测序产生的Reads过短导致的技术困难的同时,也方便利用基因组位置作为桥梁,来将测序得到的数据与前期研究产生的注释结果进行整合。Reads mapping往往被做为深度测序数据分析的第一步,其质量的好坏以及速度的快慢,都会直接对后续的分析工作产生影响。
本质上,Reads mapping还是一个双序列比对的问题。但深度测序技术的特点使之与我们之前讲到的经典序列比对问题存在着较大的不同:
1,首先是序列的长度,在之前的Needleman-Wunsch和Smith-Waterman算法中,我们都是假定用来比对的两条序列在长度上相差不大,但在Reads mapping过程中,两者的长度却有着数量级的差异:Reads的长度通常不超过100bp,而参考基因组却通常在上百Mb.
2,其次是数据量,深度测序产生的Reads动辄达到几百Gb,相当于几十个人类基因组
3,数据质量,在双序列比对中,我们通常假定输入序列本身不会有错,但目前深度测序产生的Reads质量参差不齐,错误率较高

与之前的双序列比对过程中,两条序列的“地位”对等不同,在Reads mapping过程中,相比于基因组来说,Read可以被认为是完全“嵌入”(Embedded)其中的。事实上,这里我们所需要的是一个混合了全局比对和局部比对的混合型的alignment(hybrid alignment).对于Read来说,是Global alignment;而对于基因组来说则是局部比对。于是,我们可以有一个很直观的想法,就是在之前提到的比对隐马尔可夫模型进行一些调整。例如,我们可以降低向短序列上的gap状态转移概率,甚至将相关状态直接去掉。但是这个方法虽然可以强制避免对短序列上过高的罚分,但也会最终影响比对的质量。而且,计算量也是非常大
事实上那个,我们考虑到除了少数多拷贝的基因之外,一个给定的read通常只来自基因组上的特定位置,就可以预计到从绝大部分位置其实的尝试最终都是无效的,这也导致了时间的巨大浪费。因此我们需要借鉴blast的思路。采用seeding-and-extending的策略。
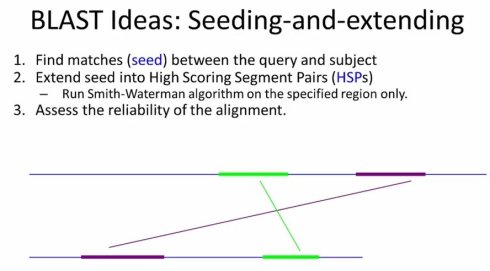
具体来说,我们首先通过对基因组建立索引,从而将Read快速定位,而后再通过标准动态规划的算法,构建最终的alignment。而所谓的索引(index)本质上是对数据的分组,基于对原始数据中的key应用索引函数(index function)处理,可以将原始数据划分为若干个更小的组,并通过将与key相关的检索限制在某个小组里来缩小搜索空间并进而降低搜索的时间。而好的索引函数应当尽力确保划分后的数据组大小相近,这样可以更好的降低平均的检索时间。
哈希(Hash)一种很常用的索引的方法,通过对原始数据中的key应用哈希函数(Hash function)计算的值作为索引地址,在理想情况下只需要常数时间,即哦O(1),即可完成数据的查找。例如,对于一段基因组序列,我们可以首先参考BLAST思路,将之切分成若干seed片段,而后针对每个seed片段计算其哈希值作为其索引表的地址,并在地址表(Address Table)中保存这段序列及其在基因组中的坐标,这样,在reads中在出现这个片段时,我们就可以在地址表中迅速地找到其在基因组上出现的位置。在Reads mapping过程中,为了提高灵敏度,通常会允许若干碱基的错配

这时,我们就可以依据抽屉原理,将read划分为若干不重叠的块。例如,我们将read划分为3个不重叠的块,就可以确保对于在错配不超过两个的情况下至少有一个片段可以作为与基因组完全匹配的种子来查询索引,从而大大提高Reads mapping的速度。因此,ELAND,SOAP1,MAQ等早期的Reads mapping算法均广泛应用了这一思路。但是我们可以很容易想到,如果在搜索的时候允许较多的错配位点,则需要将序列分解成很多小的区块。这会导致性能的急剧下降。
因此,从2009年开始,数据压缩算法中常用的前缀树(prefix tree)和后缀树(suffix tree)开始被应用于reads mapping工具的索引中。这些数据结构通过合并共享子串(shared substring)降低了内存消耗,并可以方便的实现对最长公共子串(longest substring)的查找。
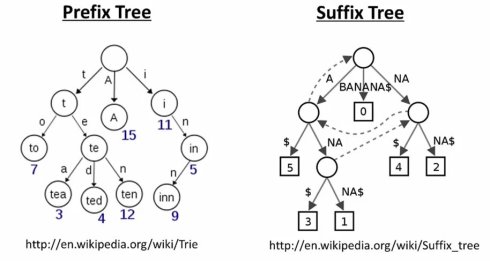
例如,目前比较流行的BOWTIE,BWA,SOAP2等工具均采用了基于后缀(suffix)的Burrows-Wheeler transform,,可以逐位对比片段进行延伸,大大提高了内存使用的效率和比对的速度。
在通过索引查找到相应种子hit之后,我们就可以参照之前BLAST的思路,向左右两个方向延伸扩展后,应用动态规划算法确定最终的比对。

但是呢,在这里我们要注意,和BLAST数据搜索不同的是,目前新一代测序技术的错误率还很高。对于一个错配位点,我们就需要考虑是测序错误引起假象(artifact)的可能性。因此,LiHeng和Richard Durbin在2008年提出了Mapping Quality分数的概念,来处理这一问题。具体来说,我们首先鉴定所有看到的错配都是由于测序错误引起的。那么此时的概率即为各个错配碱基对于的概率乘积。我们将其log-ransformation后的数值称之为SQ,在对所有可能的mapping位置都计算相应的SQ值之后,

我们就可以进一步对每个位置计算出错误mapping的概率E,值得注意的是,这里计算错误概率是同时考虑了序列相似程度和测序质量。因此在实际数据分析中,更多的使用mapping quality而非序列比对分数来筛选reads

当将测序得到的reads正确的map到参考基因组后,就可以开始鉴定遗传变异。根据遗传变异区域的尺寸,我们可以将之分为在单个碱基水平的单核苷酸变异SNV和涉及到多个碱基的结构变异SV.SNV又可以继续划分为碱基替换(也就是常说的snp)和indel。结构变异也可以划分为大尺度的插入/删除,倒转,易位,以及拷贝数变异等多种类型。在目前的遗传分析中,SNP是最常设计到遗传变异,因此我们接下来也以SNP鉴定为例,简要的介绍遗传变异鉴定的基本原理和方法。
SNP calling只是确定那个基因组位点存在变异,并不涉及对应位点的基因型,而genotype calling则是在SNP calling的基础上,进一步确定变异位点的基因型,包括是纯和的还是杂和的等等。在reads被正确map的前提下,可以直接参考特定位点上map的reads,运用经验规则(rule of thumb)来计数(counting)的方法,同时完成SNP calling与Genotyping。例如,基于测序质量去掉不可靠的碱基后,我们可以粗略的将突变位点(alternative allele)频率小于20%或大于80%的作为纯和变异位点,而将频率位于20%-80%的最为杂和变异位点。事实上,这个简单的规则在测序深度较深的纯和变异位点表现不错,但对于杂和位点及测序深度不太高的条带则差强人意;同时,这种朴素(naive)的方法严重依赖于主观选取的参数,就是我们说到的20,80,无法对结果的可靠性给出客观的指标。因此,在实践中中基于概率的SNP calling 和Genotyping模型更为常用。
下面以一个最简单的概率模型来演示。
对于一个二倍体记忆组,一个位点可能的基因型只有三种,AA,Aa和aa.其中,假定在这个位点上,我们看到了k条reads为A(big A),而(n-k)条reads为a(small A).如果真实的基因型是AA,那么这里(n-k)条的reads就必然是测序错误,那么对于的概率就是相应的n-k条reads在这个位点测序错误概率的乘积。类似的,如果真实的基因型是aa,那么对于的概率就是k条为A的reads在这个位点测序错误概率的乘积。真实的基因型为Aa时的概率就是1减去前两者的和。接下来,如果我们能够基于背景知识确定每种基因型在人群中的分布先验概率,我们就可以利用贝叶斯公式计算相应的后验概率以便进一步提高genotyping的准确性。

真实使用genotyping模型要考虑的因素远较这个大大简化了的模型(model)为多,但基本思想是类似的。
参考资料
- 魏丽萍老师的课件
