【4.5.3】同源建模--modeller
比较蛋白质结构建模主要基于给定蛋白质序列(靶标,target)与一种或多种已知结构的蛋白质(模板,templates)的比对来预测其三维结构。 预测过程包括折页分配(fold assignment),目标模板对齐(target-template alignment),模型构建(model building)和模型评估(model evaluation)。 本单元描述了:
- 如何使用程序MODELLER计算比较模型
- 如何使用此类模型的ModBase数据库,
- 并讨论了比较建模的所有四个步骤,
- 经常观察到的错误以及一些应用程序。
- 以来自阴道毛滴虫(TvLDH,Trichomonas vaginalis )的乳酸脱氢酶建模为例。
- 还介绍了MODELLER软件的下载和安装。
一、前言
蛋白质序列的功能表征是生物学中最常见的问题之一。通常,所研究蛋白质的精确三维(3-D)结构有助于完成此任务。在没有实验确定的结构的情况下,比较或同源性( comparative or homology )建模通常会为与至少一个已知蛋白质结构相关的蛋白质提供有用的3-D模型(Marti-Renom等,2000; Fiser,2004; Misura和Baker,2005; Petrey和Honig,2005; Misura等,2006)。比较模型主要基于给定蛋白质序列(靶标)与一种或多种已知结构的蛋白质(模板)的比对来预测其3-D结构。
比较建模包括四个主要步骤(Marti-Renom等,2000;图5.6.1):
- 折叠分配(fold assignment),用于确定目标与至少一个已知模板结构之间的相似性;
- 靶序列和模板的比对;
- 根据与所选模板的一致性建立模型;
- 预测模型评估。
有几种计算机程序和Web服务器可自动执行比较建模过程(表5.6.1)。 CAMEO(Haas等,2013)和半年两次的CASP(蛋白质结构预测技术的关键评估; Moult,2005; Moult等,2009)实验评估了由许多服务器计算的模型的准确性。

尽管自动化可以使专家和非专家都可以进行比较建模,但在困难情况下,通常仍需要人工干预以最大化模型的准确性。 表5.6.1中列出了许多可用于比较建模的资源。



本单元介绍如何使用程序MODELLER(基本协议)来计算比较模型。 基本协议继续讨论比较建模的所有四个步骤(图5.6.1),经常观察到的错误以及ModBase数据库和关联的Web服务。 支持协议介绍了如何下载和安装MODELLER。
二、基本流程:使用单个模型基于单个模板模拟滴虫滴虫(TvLDH,TRICHOMONAS VAGINALIS)中的乳酸脱氢酶
。。。。
三、安装
四、评论 COMMENTARY
背景资料
如前所述,比较建模包括四个主要步骤:折叠分配,目标模板对齐,模型构建和模型评估(Marti-Renom等,2000;图5.6.1)。
4.1 折叠分配和目标模板比对
尽管从逻辑上说,折叠分配和序列结构比对是比较建模过程中的两个不同步骤,但实际上,几乎所有折叠分配方法也都提供了序列结构比对。 过去,对折叠分配方法进行了优化,以提高检测远程相关同系物的灵敏度,通常是以对准精度为代价的。 但是,最近的方法同时优化了灵敏度和对准精度。 因此,在下面的讨论中,将折叠分配和序列结构比对视为一个步骤,并说明所需的差异。
折叠分配 Fold assignment
比较建模的主要要求是鉴定一种或多种与靶序列具有可检测的相似性的已知模板结构。 通过扫描结构数据库,例如PDB(Berman et al。,2000),SCOP(Andreeva et al。,2004),DALI [UNIT 5.5(Holm et al。,2006); 另请参见Dietmann等。 (2001)]和CATH(Pearl等,2005),以目标序列作为查询。 根据所使用的方法,通常根据序列同一性或统计量(例如E值或z评分)来量化检测到的相似性。
序列-结构关系的三种机制
序列-结构关系可在序列相似性谱中细分为三种不同的机制:
- 易于检测的关系,特征在于> 30%的序列同一性;
- “暮光区(twilight zone)”(Rost,1999),对应于具有统计学上显着序列相似性的关系,同一性在10%至30%范围内;
- “午夜区”(Rost,1999年),与统计上无关紧要的序列相似性相对应。
成对序列比对方法: Pairwise sequence alignment methods
对于同一性高于30%到40%的紧密相关的蛋白质序列,所有方法产生的比对几乎总是正确的。 在这种情况下搜索合适模板的最快方法是使用简单的成对序列比对方法,例如SSEARCH(Pearson,1994),BLAST(Altschul等,1997)和FASTA(Pearson,1994)。 布伦纳等(1998年)表明,这些方法在不到40%的序列同一性时只能检测到约18%的同源对,而当序列同一性在30%到40%之间时,它们可以识别出90%以上的相关性(Brenner等人, 1998)。 另一个基于200个具有0%至40%序列同一性的参考结构比对的基准表明,BLAST仅能正确比对26%的残基位置(Sauder等,2000)。
轮廓序列比对方法:Profile-sequence alignment methods
搜索的敏感性和比对的准确性随着关系进入暮光区( twilight zone)而变得越来越困难(Saqi等,1998; Rost,1999)。在这一领域的重大进步是(Gribskov等,1987)引入了图谱方法(profile methods)。序列图谱来自多个序列比对,并指定每个比对位置的残基类型出现。多序列比对中的信息通常编码为位置特异性得分矩阵(PSSM; Henikoff和Henikoff,1994; Altschul等,1997)或隐马尔可夫模型(HMM; Krogh等,1994 ; Eddy,1998)。为了识别用于比较建模的合适模板,靶序列的概况用于针对模板序列的数据库进行搜索。与基于成对序列的方法相比,轮廓序列方法在检测暮光区中的相关结构时更敏感。它们在40%序列同一性下检测到的同源物数量大约是两倍(Park等,1998; Lindahl和Elofsson,2000; Sauder等,2000)。所得的图谱-序列比对在0%至40%序列同一性范围内正确比对约43%至48%的残基(Sauder等,2000; Marti-Renom等,2004)。这个数字几乎是成对测序方法的两倍。轮廓序列比对的常用程序是PSI-BLAST(Altschul等,1997),SAM(Karplus等,1998),HMMER(Eddy,1998),HHsearch(Soding,2005),HHBlits(Remmert等) ,2012年)和BUILD_PROFILE(MODELLER的一部分; Sali和Blundell,1993年)。
谱图-谱图比对方法(Profile-profile alignment methods):
作为自然的扩展,谱-序列比对方法已导致谱-谱-比对方法,其通过针对模板谱数据库,而不是模板序列数据库扫描靶序列的谱,来搜索合适的模板结构。事实证明,这些方法包括迄今为止最敏感,最准确的折叠分配和对齐方案(Edgar和Sjolander,2004年; Marti-Renom等,2004年; Ohlson等,2004年; Wang和Dunbrack,2004年)。与谱图序列方法相比,谱图谱方法在超家族水平上可检测到多28%的关联,并将比对准确性提高15%至20%(Marti-Renom等,2004; Zhou和Zhou,2005)。轮廓-轮廓对齐方法有多种变体,它们使用的评分功能不同(Pietrokovski,1996; Rychlewski et al。,1998; Yona and Levitt,2002; Panchenko,2003; Sadreyev and Grishin,2003; von Ohsen等人,2003; Edgar,2004; Marti-Renom等,2004; Zhou和Zhou,2005)。但是,一些分析表明,这些方法的整体性能是可比的(Edgar和Sjolander,2004; Marti-Renom等,2004; Ohlson等,2004; Wang和Dunbrack,2004)。
某些可用于检测合适模板的程序:FFAS(Jaroszewski et al。,2005),SP3(Zhou and Zhou,2005),SALIGN(Marti-Renom et al。,2004),HHBlits(Remmert et al(2012年),HHsearch(Soding,2005年)和PPSCAN(MODELLER的一部分)(Sali和Blundell,1993年)。
序列结构线程化方法 Sequence-structure threading methods::
当序列同一性下降到暮光区的阈值以下时,对于上述基于序列的方法,序列或它们的谱图中通常没有足够的信号来检测真实的关系(Lindahl和Elofsson,2000)。序列结构穿线方法在这种情况下最有用,因为即使在没有任何统计学上显着的序列相似性的情况下,有时它们也可以识别常见的折叠(Godzik,2003)。这些方法通过使用从模板得出的结构信息来实现更高的灵敏度。序列结构匹配的准确性是由相应的粗略模型的评分而不是序列相似性来评估的,就像序列比较方法一样(Godzik,2003)。用于评估准确性的评分方案基于残基取代表,后者取决于结构特征,例如溶剂暴露,二级结构类型和氢键性质(Shi等人,2001; Karchin等人,2003; McGuffin和Jones,2003; Zhou and Zhou,2005)或比对所暗示的残基相互作用的统计潜力(Sippl,1990; Bowie等,1991; Sippl,1995; Skolnick和Kihara,2001; Xu等,2003) 。结构数据的使用不必限于对齐的序列-结构对的结构侧。例如,SAM-T08利用靶序列的预测局部结构来增强同源物检测和比对准确性(Karplus等,2003)。常用的线程程序是GenTHREADER(Jones,1999; McGuffin and Jones,2003),3D-PSSM(Kelley et al。,2000),FUGUE(Shi et al。,2001),SP3(Zhou and Zhou,2005),SAM -T08 multi-track(Karchin等,2003; Karplus等,2003)和MUSTER(Wu and Zhang,2008)。
迭代序列结构比对和模型构建:Iterative sequence-structure alignment and model building:
另一种策略是通过迭代计算比对,构建模型和评估模型的过程来优化比对。这样的协议可以对不具有统计意义的比对进行采样,并确定产生最佳模型的比对。尽管此过程可能很耗时,但在困难的情况下,它可以显着提高所得比较模型的准确性(John和Sali,2003年)。
精确比对的重要性: Importance of an accurate alignment
无论使用哪种方法,在序列结构关系的暮色和午夜(twilight and midnight)区域中搜索通常会导致假阴性,假阳性或包含越来越多的缺口和比对错误的比对。在这种情况下,提高方法的性能和准确性仍然是当今比较建模的主要任务之一(Moult,2005年)。必须计算目标模板对之间的精确对齐,因为比较建模几乎永远无法从对齐错误中恢复(Sanchez和Sali,1997a)。
模板选择:
在获得所有相关蛋白质结构及其与靶序列的比对的列表之后,根据比较模型的目的确定模板结构的优先级。 模板结构可以仅基于靶标模板序列同一性或几种其他标准的组合来选择,例如结构的实验准确性(X射线结构的分辨率,NMR结构每个残基的束缚数),保守性 活性位点残基,已结合感兴趣的配体的完整结构以及与溶剂,pH和四级接触有关的现有生物学信息。 不必只选择一个模板。 实际上,使用几个与靶序列等距的模板通常会提高模型的准确性(Srinivasan和Blundell,1993; Sanchez和Sali,1997b)。
4.2 Model building
通过组装刚体来建模:Modeling by assembly of rigid bodies:
比较模型中的第一个且仍被广泛使用的方法是,通过从对齐的蛋白质结构中获得的少量刚体组装模型(Browne等人,1969; Greer,1981; Blundell等人(1987年)。该方法基于将蛋白质结构自然分解为保守的核心区域,连接它们的可变环以及装饰主链的侧链。例如,以下半自动化过程在计算机程序COMPOSER中实现(Sutcliffe等,1987)。
- 首先,选择并叠合(superposed)模板结构。
- 其次,通过对模板结构中结构保守区域的Cα原子的坐标求平均值来计算“框架”。
- 第三,目标模型中每个核心区域的主链原子,是通过将核心片段从序列最接近目标的模板上叠加到框架上而获得的。
- 第四,通过扫描所有已知蛋白质结构的数据库以识别适合锚定核心区域并具有兼容序列的结构可变区来生成环(Topham等,1993)。
- 第五,基于侧链的固有构象偏好和模板结构中等效侧链的构象对侧链进行建模(Sutcliffe et al。,1987)。
- 最后,通过限制能量最小化或分子动力学改进来改善模型的立体化学。
当使用多个模板结构来构建框架时,以及使用与模板序列与目标序列相似性相对应的权重将模板平均化到框架中时,可以提高模型的准确性(Srinivasan和Blundell,1993)。刚体组装可能会在将来对建模进行改进,包括引入刚体位移,例如螺旋和β片的堆积的相对位移(Nagarajaram等,1999)。实现此方法的其他三个程序是3D-JIGSAW(Bates等,2001),RosettaCM(Song等,2013)和SWISS-MODEL(Schwede等,2003)。
通过片段匹配或坐标重建进行建模:Modeling by segment matching or coordinate reconstruction
通过坐标重建进行建模的基础是发现蛋白质结构的大多数六肽片段只能聚类为100个结构上不同的类别(Jones和Thirup,1986; Claessens等,1989; Unger等)等,1989; Levitt,1992; Bystroff和Baker,1998)。因此,可以通过使用来自模板结构的原子位置的子集作为引导位置来构建比较模型,以识别和组装适合这些引导位置的短的全原子片段。指导位置通常对应于在模板结构和靶序列之间的比对中保守的区段的Cα原子。可以通过扫描所有已知的蛋白质结构(包括与正在建模的序列无关的蛋白质结构)来获得适合引导位置的所有原子片段(Claessens等,1989; Holm和Sander,1991)。构象搜索受到能量函数的限制(Bruccoleri和Karplus,1987; van Gelder等,1994)。该方法既可以构建主链原子也可以构建侧链原子,并且还可以对未对齐的区域(间隙)进行建模。它在程序SegMod中实现(Levitt,1992)。甚至某些侧链建模方法(Chinea等,1995)和基于在已知结构的数据库中找到合适片段的环构建方法的类型(Jones和Thirup,1986)也可以看作是段匹配或坐标重建方法。 。
通过满足空间限制条件进行建模:Modeling by satisfaction of spatial restraints:
此类方法首先以对目标序列的结构与相关蛋白质结构的比对为指导,对目标序列的结构产生许多限制或约束。该程序从概念上讲类似于从NMR衍生的约束中确定蛋白质结构所使用的程序。通常通过假设模板中的对齐残基与目标结构之间的对应距离相似来获得约束。这些同源性的约束条件通常由对键长,键角,二面角和从分子力学力场获得的非键原子原子接触的立体化学约束所补充。然后,通过最小化所有约束的违反来导出模型。该优化可以通过距离几何或空间优化来实现。例如,一种优雅的距离几何方法从距离和二面角的上下边界构造所有原子模型(Havel和Snow,1991)。
4.3 Comparative protein structure modeling by MODELLER:
MODELLER,开发者自己的比较模型程序属于这一组方法(Sali和Blundell,1993; Sali和Overington,1994; Fiser等,2000; Fiser等,2002)。 MODELLER通过满足空间约束来实现比较蛋白质结构建模。 该程序旨在使用尽可能多的有关目标序列的信息。
同源性约束:Homology-derived restraints
在模型构建的第一步中,目标序列的距离和二面角约束来自其与模板3-D结构的比对。这些限制的形式是从对相似蛋白质结构之间关系的统计分析中获得的。该分析依赖于105个家族比对数据库,其中包括416种具有已知3-D结构的蛋白质(Sali和Overington,1994)。通过扫描比对数据库,获得了量化各种相关性的表格,例如来自两个相关蛋白的两个等效Cα-Cα距离之间或等效主链二面角之间的相关性(Sali和Blundell,1993)。这些关系表示为条件概率密度函数(pdf,probability density functions ),可以直接用作空间约束。例如,根据所考虑的残基类型,等效残基的主链构象以及两种蛋白质之间的序列相似性,计算出不同主链二面角值的概率。另一个示例是给定两个相关蛋白质结构中等效距离的pdf,其中包含特定Cα-Cα距离。该方法的一个重要特征是从蛋白质结构比对数据库中凭经验获得空间约束的形式。
立体化学约束 Stereochemical restraints
在第二步中,将空间约束和CHARMM22力场项强制执行适当的立体化学(MacKerell等,1998),将其组合为目标函数。目标函数的一般形式类似于分子动力学程序中的形式,例如CHARMM22(MacKerell等,1998)。目标函数取决于形成建模分子的约10,000个原子(3-D点)的笛卡尔坐标。对于一个10,000原子的系统,大约有200,000个约束。每个术语的功能形式都很简单;它包括一个二次函数,谐波上下界,余弦,几个高斯函数的加权和,库仑定律,伦纳德·琼斯势能(Lennard-Jones potential)和三次样条(cubic splines)。当前的几何特征包括分别在两个,三个,四个和八个原子之间的距离,角度,二面角,一对二面角,距离集合中的最短距离,溶剂可及性和原子密度表示为中心原子周围的原子数。可以使用某些约束来约束伪原子,例如,几个原子的重心。
目标函数的优化
最后,通过在笛卡尔空间中优化目标函数获得模型。 通过使用可变目标函数方法(Braun和Go,1985),采用共轭梯度和分子动力学模拟退火方法(Clore等,1986)进行了优化。 可以通过改变初始结构来计算几个略有不同的模型,并且这些模型之间的可变性可以用来估计折痕对应区域中的误差的下限。
从实验数据得出的约束条件
由于通过满足空间约束条件进行建模可以使用许多有关目标序列的信息,因此,这可能是所有比较建模技术中最有前途的。通过满足空间约束进行建模的优势之一是,可以将源自许多不同来源的约束轻松添加到同源性约束中。例如,可以通过二级结构堆积的规则(Cohen等,1989),疏水性分析(Aszodi和Taylor,1994)和相关的突变(Taylor等,1994),平均力的经验潜力来提供约束 (Sippl,1990),核磁共振(NMR)实验(Sutcliffe等,1992),交联实验,荧光光谱,电子显微镜中的图像重建,定点诱变(Boissel等,1993)和直觉,以及其他来源。特别是在困难的情况下,可以通过使比较模型与可用的实验数据和/或有关蛋白质结构的更一般的知识一致来改进它。
相对准确性,灵活性和自动化性
最优使用时,各种模型构建方法的准确性相对相似(Marti-Renom等,2002)。 诸如模板选择和比对准确性之类的其他因素通常对模型准确性有较大影响,尤其是对于基于与模板的低序列同一性的模型而言。 但是,重要的是,建模方法必须具有一定程度的灵活性和自动化功能,以便更轻松,更快速地获得更好的模型。 例如,当对路线进行更改时,一种方法应允许轻松地重新计算模型。 它也应该足够直接,即可基于多个模板计算模型,并应提供工具以整合有关目标的先验知识(例如,交联约束,预测的二级结构),并允许从头开始对插入物进行建模(例如,循环 loops) ,这对于功能注释至关重要。
4.4 Loop modeling:
Loop建模是30%到50%序列同一性范围内比较建模的一个特别重要的方面。在总体相似性的这一范围内,同系物之间的环变化,而核心区域仍相对保守且精确对齐。环通常在定义给定蛋白质的功能特异性,形成活性和结合位点方面起重要作用。环路建模可以看作是一个微小的蛋白质折叠问题,因为多肽链给定片段的正确构象必须主要从片段本身的序列中计算出来。但是,Loop通常太短,无法提供有关其局部折叠的足够信息。即使不同蛋白质中的相同十肽也不总是具有相同的构象(Kabsch和Sander,1984; Mezei,1998)。跨越环的核心锚定区域和支撑环的其余蛋白质的结构提供了一些其他限制。尽管已经描述了许多环建模方法,但是正确而可靠地对大于约10至12个残基的环建模仍然具有挑战性(Fiser等,2000; Jacobson等,2004; Zhu等,2006)。
Loop建模方法主要有两类:
- 扫描所有已知蛋白质结构数据库的数据库搜索方法,以找到适合锚定核心区域的片段(Jones和Thirup,1986; Chothia和Lesk,1987);
- 依赖于优化评分功能的构象搜索方法(Moult和James,1986; Bruccoleri和Karplus,1987; Shenkin等,1987)。
也有一些方法将这两种方法结合在一起(van Vlijmen和Karplus,1997; Deane和Blundell,2001)。
通过数据库搜索进行Loop建模:
当创建特定Loop的数据库来解决同一类环路的建模问题(例如β-发夹,β-hairpins)时,采用数据库搜索方法进行环路建模是准确而高效的(Sibanda等,1989),或在特定折叠上形成环,例如免疫球蛋白折叠中的高变区(Chothia and Lesk,1987; Chothia et al。,1989)。有尝试将环构象分类为更一般的类别,从而扩展了数据库搜索方法的适用性(Ring等,1992; Oliva等,1997; Rufino等,1997; Fernandez-Fuentes和Fiser,2006 )。但是,数据库方法受到限制,因为可能的构象数量随环的长度呈指数增加,并且直到1990年代后期,只能使用已知蛋白质结构的数据库对长达7个残基的环进行建模(Fidelis等人, 1994; Lessel和Schomburg,1994)。但是,近年来PDB的增长在很大程度上消除了这个问题(Fernandez-Fuentes和Fiser,2006)。
通过构象搜索进行Loop建模:
有许多此类方法,它们利用不同的蛋白质表示,目标函数以及优化或枚举算法。搜索算法包括最小摄动法(minimum perturbation method)(Fine等人,1986),通过旋转异构体库进行二面角搜索(Zhu等人,2006; Sellers等人,2008),分子动力学模拟(Bruccoleri和Karplus,1990; van Vlijmen和Karplus,1997),遗传算法(Ring等,1993),蒙特卡洛和模拟退火(Higo等,1992; Collura等,1993; Abagyan和Totrov,1994),多副本同时搜索(multiple copy simultaneous search)(Zheng等,1993),自洽场优化( self-consistent field optimization )(Koehl和Delarue,1995),机器人启发的运动学闭合(Mandell等,2009)以及基于图论的枚举(Samudrala和Moult,1998)。通过对采样的loop构象进行聚类并部分考虑熵对自由能的贡献,可以进一步提高loop预测的准确性(Xiang等,2002)。提高定量环预测准确性的另一种方法是考虑溶剂效应。隐式溶剂化模型的改进(例如广义Born溶剂化模型)促使他们在loop建模中使用。可以将溶剂对自由能的贡献添加到评分函数中进行优化,也可以在生成不包含溶剂项的评分函数后将其用于对采样的环构象进行排序(Fiser等,2000)。 ; Felts等,2002; de Bakker等,2003; DePristo等,2003)。
4.5 通过迭代比对,模型构建和模型评估来建立比较模型
比较模型或同源蛋白质结构建模受到建模序列与已知三维结构的相关蛋白质比对中的错误的严重限制。为了改善这个问题,可以使用一种迭代方法来优化比对和它所隐含的模型(Sanchez和Sali,1997a; Miwa等,1999)。这项任务可以通过遗传算法协议来完成,该协议从一组初始比对开始,然后通过重新比对,模型构建和模型评估进行迭代,以优化模型评估得分(John和Sali,2003年)。在此迭代过程中:(1)通过使用许多遗传算法运算符来构建新的比对,例如比对突变和交叉; (2)通过满足空间约束来建立对应于这些路线的比较模型,如程序MODELLER中所实现的; (3)通过综合评分评估模型,部分取决于原子统计潜力(Melo等,2002)。在非常困难的19个建模目标集上测试该程序时,这些目标与它们的模板结构仅共享4%至27%的序列同一性,相对于初始比对,平均最终比对准确度从37%增至45%(已测量比对准确度表示与参照结构为基础的比对相同的位置)。相应地,平均模型精度从43%增加到54%(模型精度是通过模型的Cα原子在叠加的天然结构中与相应Cα原子相距5Å以内的百分比来衡量的)。
4.6 比较模型中的错误 Errors in comparative models
随着目标和模板之间的相似度降低,模型中的错误也会增加。 比较模型中的错误可以分为五类(Sanchez和Sali,1997a,1997b;图5.6.14),如下所示:
侧链堆积的错误(图5.6.14A):
随着序列的不同,蛋白质核心中侧链的堆积也会发生变化。 有时甚至连相同侧链的构象也无法保留,这是许多比较建模方法的陷阱。 如果侧链错误发生在涉及蛋白质功能的区域,例如活性位点和配体结合位点,则至关重要。

正确对齐区域中的扭曲和移位(图5.6.14B) Distortions and shifts in correctly aligned regions
由于序列差异,即使整体折叠保持不变,主链构象也会发生变化。 因此,有可能在模型的某些正确对齐的部分中,模板与目标局部不同(> 3Å),从而导致该区域出现错误。 结构差异有时不是由于序列差异引起的,而是结构确定或结构确定在不同环境中的伪像的结果(例如,晶体中亚基的堆积)。 同时使用多个模板可以最大程度地减少此类错误(Srinivasan和Blundell,1993; Sanchez和Sali,1997a,1997b)。
没有模板的区域中的错误(图5.6.14C)
在模板结构中没有等效区域(即插入或环)的目标序列片段是最难建模的区域。 如果插入相对较短,长度小于9个残基,则某些方法可以正确预测骨架的构象(van Vlijmen和Karplus,1997; Fiser等,2000; Jacobson等,2004)。 成功预测的条件是正确的对齐方式和围绕插入物的准确建模环境。
由于比对导致的错误(图5.6.14D)
比较模型中最大的单一错误源是比对,尤其是当目标模板序列同一性降低到30%以下时。 然而,可以通过两种方式使对准误差最小化。 首先,即使这些序列中的大多数不具有已知结构,通常也可以使用大量序列来构建多重比对。 多重比对通常比成对比对更可靠(Barton和Sternberg,1987; Taylor等,1994)。 改善比对的第二种方法是迭代修改比对中与模型中预测误差相对应的区域(Sanchez和Sali,1997a,1997b; John和Sali,2003)。
不正确的模板(图5.6.14E)
当使用远距离相关的蛋白质作为模板时(即,序列同一性<25%),这是一个潜在的问题。 很难区分基于不正确模板的模型和基于与正确模板的不正确对齐的模型。 在这两种情况下,评估方法都将预测不可靠的模型。 靶序列中关键功能或结构残基的保守性增加了给定倍数分配的置信度。
4.7 预测模型的准确性
预测模型的准确性决定了可以从中提取的信息。 因此,在缺乏已知结构的情况下估算模型的准确性对于解释模型至关重要。
折叠的初步评估 Initial assessment of the fold:
如前所述,使用共享超过30%序列同一性的模板结构计算的模型表明总体准确的结构。但是,当序列同一性较低时,模型评估的第一方面是确认是否使用正确的模板进行建模。在这种情况下操作时,折返分配步骤通常只会产生假阳性。进一步的复杂性是,在如此低的相似度下,比对通常包含许多错误,使得难以区分一方面的错误模板和另一方面与正确的模板的错误对齐。有几种利用3-D分布图和统计潜力的方法(Sippl,1990; Luthy等,1992; Melo等,2002),通过评估其中每个残基的环境来评估序列和建模结构之间的相容性。相对于预期的环境的模型,该模型是在本机高分辨率实验结构中发现的。这些方法可用于评估建模是否使用了正确的模板。它们包括VERIFY3D(Luthy等,1992),Prosa,2003(Sippl,1993; Wiederstein和Sippl,2007),HARMONY(Topham等,1994),ANOLEA(Melo和Feytmans,1998),DFIRE(Zhou和Zhou,2002),DOPE(Shen and Sali,2006),SOAP(Dong等,2013),QMEAN local(Benkert等,2011)和TSVMod(Eramian等,2008)。
即使当模型基于具有> 30%序列同一性的比对时,包括环境在内的其他因素也会强烈影响模型的准确性。 例如,某些钙结合蛋白在与钙结合时会发生较大的构象变化。 如果使用无钙模板来模拟目标的钙结合状态,则不管目标模板与模板结构的相似性或准确性如何,该模型都是不正确的(Pawlowski等,1996)。
自我一致性评估
还应该对模型进行自我一致性评估,以确保模型满足计算时所使用的约束条件。 此外,可以使用PROCHECK(Laskowski等,1993)和WHATCHECK(Hooft等,例如,Professionals,Inc.,2002)来评估模型的立体化学(例如,键长,键角,主链扭转角和非键接触,1996)。 尽管立体化学中的错误比统计电位检测到的错误少见且信息量少,但是一组立体化学错误可能表明该区域存在较大的错误(例如,对齐错误)。
4.8 应用程序 Applications
为了最大程度地发挥比较模型的影响,应清楚地描述用于生成比较模型的建模过程,例如,通过发布或标识与此处描述的协议类似的协议。该协议还应包括使用当前可用的最佳计算工具进行的评估,例如本文中介绍的模型评估方法,以及在蛋白质模型门户网站( http://proteinmodelportal.org ; Arnold等,2009; Haas等人,2013年)。最后,该研究应合理化现有的实验数据并做出可检验的预测。例如:
- 比较模型可以帮助设计突变体以测试有关蛋白质功能的假设(Wu等,1999; Vernal等,2002);
- 在鉴定活性和结合位点时(Sheng等,1996);
- 在寻找,设计和提高给定结合位点的配体结合强度方面(Ring等人,1993; Li等人,1996; Selzer等人,1997; Enyedy等人,2001; Que等人, 2002);
- 模拟底物特异性(Xu et al。,1996);在
- 预测抗原表位中(Sali和Blundell,1993);
- 模拟蛋白质-蛋白质对接(Vakser,1995);
- 通过计算蛋白质周围的静电势来推断功能(Matsumoto等,1995);
- 在X射线结构测定中促进分子置换(Howell等,1992);
- 在基于NMR约束的精炼模型中(Modi等,1996);
- 在测试和改善序列结构比对中(Wolf等,1998);
- 注释单核苷酸多态性(Mirkovic et al。,2004; Karchin et al。,2005);
- 通过与低分辨率的冷冻电子密度图对接来对大型配合物进行结构表征(Spahn等,2001; Gao等,2003)。并合理化已知的实验观察结果。
幸运的是,如上面列出的应用所示,3-D模型不一定对生物学有帮助是绝对完美的。 可以使用特定模型解决的问题的类型确实取决于其准确性(图5.6.15)。
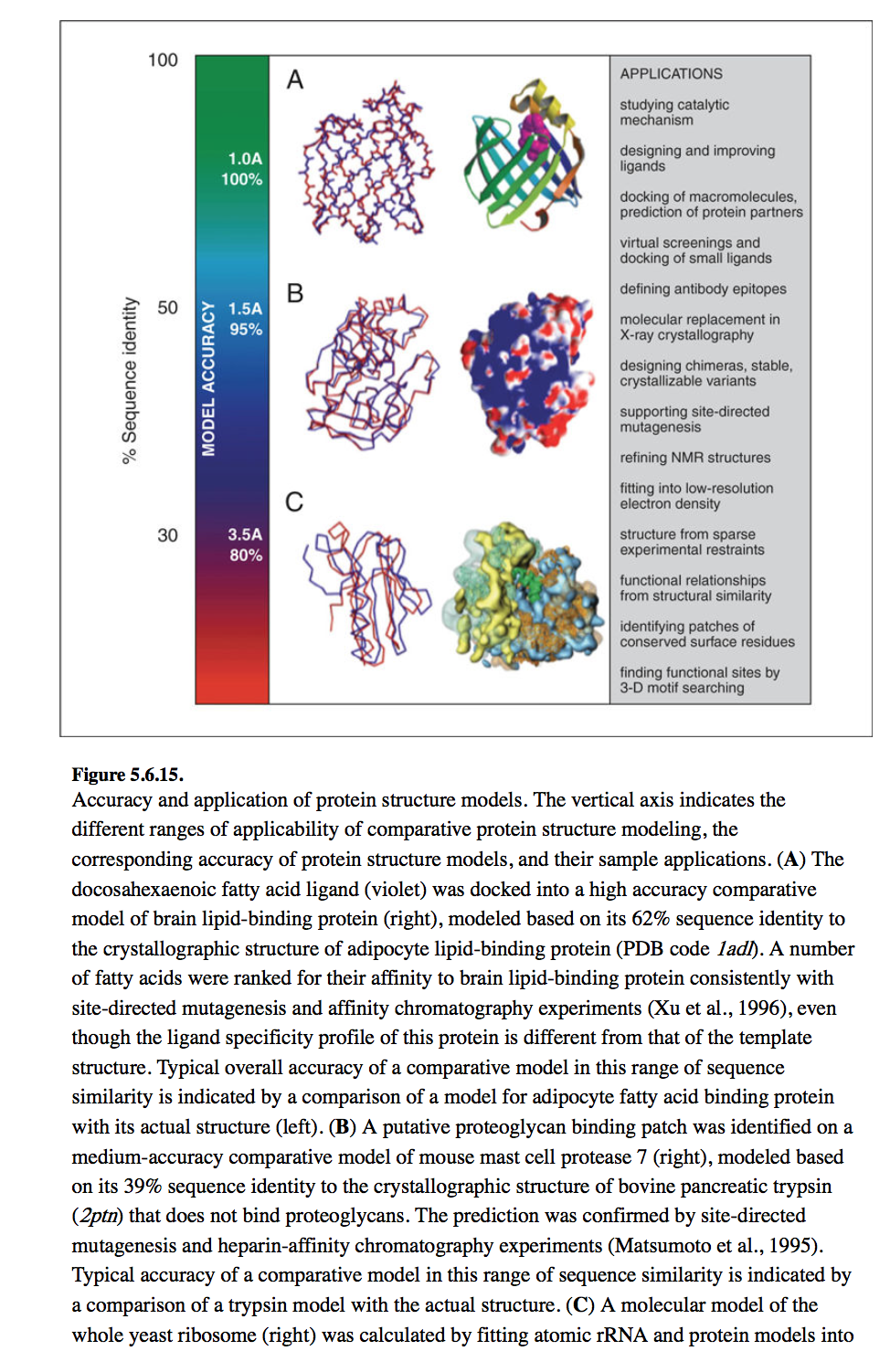

在准确性谱的低端,有些模型是基于少于25%的序列同一性的,并且有时在其正确位置的3.5Å之内有少于50%的Cα原子。 但是,此类模型可能仍具有正确的折叠,甚至有时仅知道蛋白质的折叠有时就足以预测其近似的生化功能。 这种低准确度范围内的模型与模型评估相结合,可用于确认或拒绝远距离相关蛋白之间的匹配(Sanchez和Sali,1997a,1998)。
在准确性谱的中间是基于大约35%序列同一性的模型,对应于在其正确位置3.5AÂ内建模的Cα原子的85%。幸运的是,活性和结合位点通常比其余折叠更保守,因此可以更精确地建模(Sanchez和Sali,1998)。通常,中等分辨率模型经常允许仅基于序列来完善功能预测,因为配体结合最直接地由结合位点的结构而不是其序列决定。通常可以正确预测靶蛋白在模板结构中不存在的重要特征。例如,可以从带电残基的簇中预测结合位点的位置(Matsumoto等,1995),可以从结合位点裂隙的体积预测配体的大小(Xu等, 1996)。中分辨率模型也可用于构建具有改变或破坏的结合能力的定点突变体,这反过来又可以检验关于序列-结构-功能关系的假设。中分辨率比较模型可以解决的其他问题包括设计结构紧凑,没有长尾巴,环和暴露的疏水残基的蛋白质,以使其更好地结晶,或者设计具有添加的二硫键的蛋白质,以提高稳定性。
精度谱的高端对应于基于50%或更高序列同一性的模型。 这些模型的平均准确度接近低分辨率X射线结构(3 A?分辨率)或中分辨率NMR结构(每个残基10个距离约束; Sanchez和Sali,1997b)。 这些模型所基于的路线通常几乎没有错误。 事实证明,具有如此高精确度的模型甚至对于通过分子置换方法提炼晶体结构也是有用的(Howell等,1992; Baker和Sali,2001; Jones,2001; Claude等,2004; Schwarzenbacher等) (2004年)。
良好的比较建模研究倾向于包含用于模型推导,评估和/或解释的新颖实验数据。但是,在某些情况下,只有模型,合理化和/或预测(即使没有新的实验数据)也可能会产生影响,从而加速由数据生成,解释,建模和假设构成的科学循环。
4.9 结论
在过去的几年中,比较模型的准确性和可以用、有用的准确性进行建模的蛋白质序列的比例都在逐步提高(Marti-Renom等,2000; Baker和Sali,2001; 2001 Pieper等,2014)。折叠分配,对齐以及侧链和环的建模中的错误幅度已大大降低。这些改进是更好的技术以及大量已知蛋白质序列和结构的结果。但是,所有错误仍然很明显,需要未来的方法改进。另外,迫切需要对畸变和刚体位移(distortions and rigid-body shifts)进行更精确的建模,以及检测给定蛋白质结构模型中的错误。错误检测对于模型的细化和解释都是有用的。
参考资料
- Published in final edited form as: Curr Protoc Bioinformatics. ; 54: 5.6.1–5.6.37. doi:10.1002/cpbi.3. Comparative Protein Structure Modeling Using MODELLER
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5031415/
