【4.7.3.1】结构比较--Prosa z-score
官网: https://www.came.sbg.ac.at/prosa.php
- knowledge-based potentials of mean force to evaluate model accuracy
- All calculations are carried out with Cα potentials
- Z分数严重偏离数据库平均值的可溶性球状蛋白的结构异常,并且经常发现这种结构是错误的。 对于包含跨膜区域的蛋白质,相对于数据库平均值的偏差的重要性尚不清楚。
结构生物学中的一个主要问题是蛋白质结构的实验模型和理论模型中错误的识别。 ProSA程序(Protein Structure Analysis)是一种已建立的工具,具有大量的用户基础,并经常用于实验蛋白质结构的细化和验证以及结构预测和建模。蛋白质结构的分析通常是一项艰巨而繁琐的工作。此处提供的新服务是对经典ProSA程序的直接简单易用的扩展,它利用了基于交互式Web的应用程序的优势来显示得分和能谱,从而突出了蛋白质结构中发现的潜在问题。特别是,在所有已知蛋白质结构的背景下显示蛋白质的质量得分,并在3D分子查看器中显示并突出显示结构的问题部分。该服务专门解决了从X射线分析,NMR光谱和理论计算获得的蛋白质结构验证中遇到的需求。
z得分( z-score )表示总体模型质量,并测量结构的总能量相对于从随机构象得出的能量分布的偏差(3,15)。 天然蛋白特征范围之外的Z分数表明结构错误。 为了便于解释指定蛋白质的z分数,在包含当前PDB中所有实验确定的蛋白质链的z分数的图中显示其特定值(示例如图1A所示)。 来自不同来源(X射线,NMR)的结构组具有不同的颜色。 该图可用于检查所讨论蛋白质的z分数是否在通常为属于这些组之一的相似大小蛋白质的分数范围内。
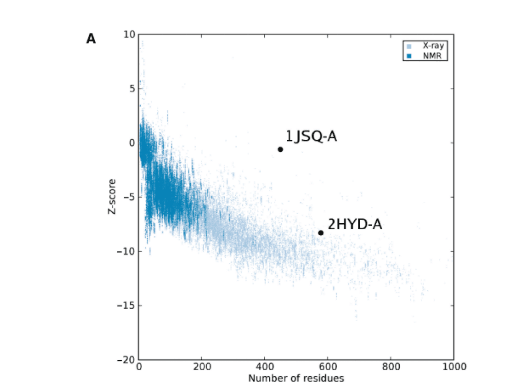
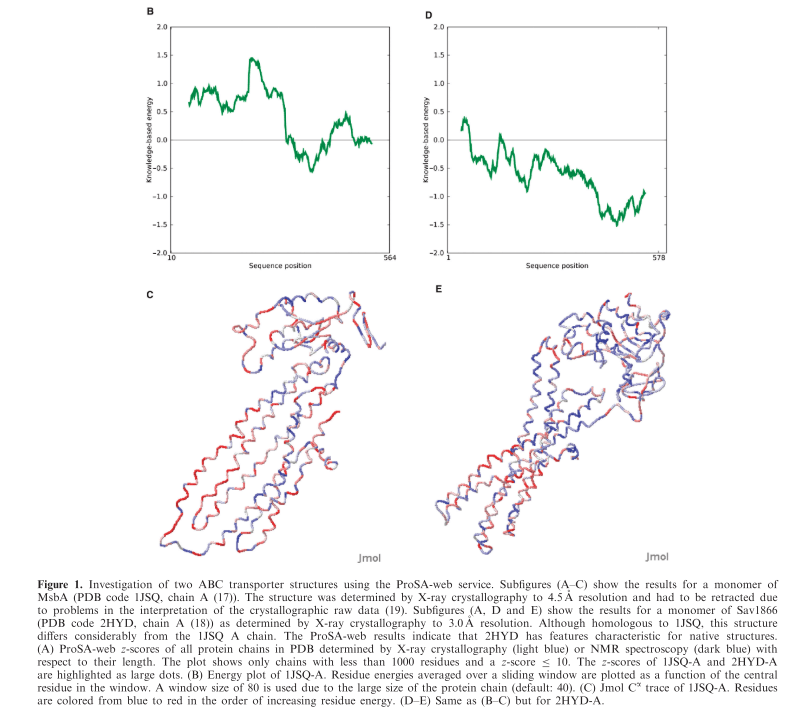
图1. 通过X射线晶体学(浅蓝色)或NMR光谱法(深蓝色)确定PDB中所有蛋白质链的ProSA-web z分数,以其长度表示。 该图仅显示了残基少于1000个且z分数≤10的链。1JSQ-A和2HYD-A的z分数突出显示为大点
在这里,我们提供了一个已公开结构(1JSQ)的示例,该结构被认为是不正确的,随后对相关蛋白进行了独立的X射线分析,得出了完全不同的构象。 与PDB中的所有结构相比,针对1JSQ获得的ProSA-web结果显示出极大的偏差(图1A)。 相反,从相关的2HYD结构获得的分数接近数据库平均值。 结果表明,对于膜蛋白,与正常值的较大偏差也可能表明结构错误
一、 prosa z-score
我们开发蛋白质溶剂系统力场的方法是基于Boltzmann原理。通过实验方法求解的三维结构集包含有关稳定溶液中自然折叠的力场的大量信息。使用波尔兹曼(Boltzmann)原理,这些力以平均力势的形式从已知结构的数据库中提取。然后,通过将这些电位作为氨基酸序列的函数进行重组,获得结构已知或未知的特定蛋白质的力场。最近已经探索了这种方法在蛋白质折叠中的适用性,并且平均场已成功应用于结构生物学中的许多问题。一个最重要的特征是力场能够区分自然折叠与折叠错误。此功能是关键,它可以识别不正确的结构确定因素。具体而言,我们对由于紧密接触或其他违反基本空间原理而引起的问题不感兴趣,而是对蛋白质链在三个维度上的正确排列感兴趣。
所用的平均场由平均力势构成,该势能根据与特定氨基酸相关的两个原子的空间分离来模拟分子内对相互作用的能量特征。在给定的结构中,沿着链的位置i和j的氨基酸残基之间的相互作用能eij,是相应残基原子之间的相互作用能之和。
在下面给出的计算中,低eij对应于Ca-Ca或Cb-Cb相互作用。从所有成对相互作用的总和中获得总相互作用能E, $ E = \frac{1}{2}\sum_{ij}e_{ij}$。 蛋白质的总能量是其序列S和构象C的函数,用符号$ E_{S,C}$表示。对应于序列S的自然折叠将用N表示。从实验或建模研究得出的模型折叠将用不同的符号X表示,因为自然折叠和观察到的折叠不一定相同。
如果X对应于(或至少与之相关)序列S的自然折叠N,则与一大组替代构象相比,$ E_{S,X}$能量应具有其最低值。可以在简单的计算机实验中测试此功能。实验确定的折叠X的骨架隐藏在大量的非天然引诱物C中。仅使用骨架原子(包括Cb)时,氨基酸序列无法从其余支架中衍生出来。现在的任务是寻找X,其中将从力场计算出的能量用作指导原理。如果与所有替代方案相比,序列S与观察到的折叠X结合在一起时能量最低,则任务成功解决。
隐藏和查找(Hide and seek)是对由一组已知的三维结构构成的多蛋白操作。这些结构使用数据库中蛋白的短片段连接在一起,要求沿着多蛋白和接头区域的局部几何形状不违反基本的空间位阻原理。这样可以确保从多蛋白中提取的任何片段都符合合理的构象。本研究中使用的多蛋白由230种已知结构的蛋白组成,总长度约为L ≈ 50,000个残基。
在“捉迷藏”(hide and seek)实验中,序列S沿着多蛋白质,移位长度为l的氨基酸,并且在每个位置C处评估了平均力能E(S,C)。
遇到的构象C = 1,…,L-l+1表示 实验中可访问的序列S的构象空间。 由于l«L,因此可用构象的数目实际上与l无关,并且接近L = 50,000。 能量E(S,C)用平均场表示序列结构对(S,C)的适合度。 这些能量通过 $z_{s,c} = (E_{S,C} - E_{s})/ \sigma_{S}$变换为z分数,其中$E_{S} = \sum_{C}E_{S,C}/(L-l+1) $ ;$\sigma_{S}$是相关的标准偏差。 特别是 $z_{s,c}$ 是目标折叠X的z得分。
如果$ E_{S,X} < E_{S,C} $且等效地 $ z_{S,X} < z_{S,C} $ ,则对于所有C≠X,成功鉴定出目标折痕X。
$z_{s,x} $可以解释为预测功效的度量 ,关于观察到的结构为X的序列S的蛋白质的力场的变化。
在这个捉迷藏(hide and seek )实验中,我们使用假设:目标构象X对应于S的自然折叠N,目的是验证这一假设。 通过将序列S分配给任意折叠X来获得故意错折叠的蛋白质。在这里,我们再次使用这样的假设,即这些折叠对相关序列最有利。 对于蛋白质错误折叠的情况,假设当然是错误的,我们期望在这些情况下具有较高的能量和较低的分数。
参考资料
- Wiederstein & Sippl (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins.
- Nucleic Acids Research 35, W407-W410. [view] Sippl, M.J. (1993) Recognition of Errors in Three-Dimensional Structures of Proteins. Proteins 17, 355-362. [view] https://academic.oup.com/nar/article/35/suppl_2/W407/2920938
