【6.2.2】基于序列和结构设计来提高蛋白表达量和稳定性(PROSS)
PROSS(Protein Repair One Stop Shop)这个方法的思路概括出来就四点:
- non-functional regions ( 8 Å away from legend)
- evolutionarily ‘‘allowed’’ sequence space (PSSM scores ≥0)
- △△Gcalc ≤-0.45 Rosetta energy units
- many mutations improve high stability and expression
前两点是为了不影响功能的情况下,提高突变的可信度,同时减少突变的数据,减少计算量。第三点是判决条件
在异源过表达(heterologous overexpression)时,许多蛋白质错误折叠或聚集,从而导致低功能产率。人类乙酰胆碱酯酶(hAChE)是一种介导突触传递的酶,是人类蛋白质的典型案例,需要乳腺系统才能获得功能性表达。我们开发了一种计算策略,设计了一种带有51个突变的AChE变体,可以改善核心堆积(core packing),表面极性( surface polarity)和骨架刚性(backbone rigidity)。与野生型hAChE相比,该变体在大肠杆菌中的表达水平高2,000倍,并且表现出高20℃的热稳定性,酶学性质没有变化,或者通过结晶学确定的活性位点构型。为了展示广泛的用途,我们同样设计了四种其他人类和细菌蛋白质。每种蛋白质测试至多三种设计,我们在大肠杆菌中获得增强的稳定性和/或更高的可溶性和活性蛋白质产量。我们的算法仅需要3D结构和数十种天然存在的同源物序列,可在 http://pross.weizmann.ac.il 获得。
一、前言
大多数天然蛋白质仅略微稳定(Magliery,2015)。 因此,从它们的自然环境中,通过过度表达,异源表达或环境条件的变化,许多蛋白质错误折叠和聚集。 过度表达挑战的最常见起源是蛋白质天然功能状态相对于其他非功能性或聚集倾向状态的稳定性较低。 通过设计具有更有利的天然状态能量的变体,可以显着增加通过异源过表达获得的可溶性和功能性蛋白质的产量,以及其他优点,例如更长的储存和使用寿命以及增强的工程潜力。
工程稳定的蛋白质变体是一个广泛追求的目标。基于系统发育分析的方法(Lehmann等,2000; Steipe等,1994)和基于结构的理性或计算设计(Borgo和Havranek,2012; Jacak等,2012; Korkegian等,2005) ; Lawrence等,2007)产生了具有改善的稳定性和更高功能表达的蛋白质(Mag- liery,2015)。然而,单个突变对稳定性贡献很小(通常%1kcal / mol)(Zhao和Arnold,1999),而稳定大和表达不良的蛋白质通常需要许多突变。然而,由于即使是一个严重破坏稳定的突变也会破坏所有其他突变带来的好处,因此高预测准确性至关重要。尽管准确性有所提高,但现有方法无意中引入破坏性突变的可能性相对较高(假阳性预测)(Borgo和Havranek,2012; Mag- liery,2015; Potapov等,2009)。因此,已发表的稳定大蛋白质的努力要么在每个实验步骤中仅包含少数预测的稳定突变(通常为%4),要么使用文库方法来鉴定稳定突变的最佳组合(Khersonsky等,2011; Sullivan等,2012; Trudeau等,2014; Whitehead等,2012; Wijma等,2014)。对于没有已建立的中到高通量筛选的蛋白质,这种方法是费力且不切实际的,更不用说具有未知功能的蛋白质。为了解决缺乏计算设计背景的广泛研究人员对稳定大型顽固蛋白质的需求,我们开发了一种基于原子Rosetta建模和系统发育序列信息的自动化算法。我们特别致力于开发一种最小化假阳性预测的通用方法,以确保只需少量变量进行实验测试即可获得高功能产量(理想情况下,只需一种变体)。我们将该算法应用于四种不同的酶和一种功能未知的蛋白质。在每种情况下,最多设计五种变体作为默认输出,相对于野生型,编码从9到67个突变。这些变体表现出增强的细菌表达产量和稳定性,而不会牺牲或改变活性。
二、设计 (计算突变扫描可最大限度地减少假阳性突变)
为了解决设计具有大面积稳定效应的变体的挑战,但是在不修改其功能的情况下,我们的计算过程始于扫描自然序列多样性。对于每个目标野生型序列,我们生成了序列比对,我们从中计算了位置特异性置换矩阵(position-spe- cific substitution matrix ,PSSM)(Altschul等,2009); PSSM代表观察每个位置的20个氨基酸中的任何一个的对数似然性。在每个氨基酸位置,“允许的”突变被定义为具有有利的PSSM评分(≥0)的那些。通过比对扫描限制允许的序列空间的基本原理是,通常通过自然选择清除有害突变;然而,扫描不是绝对的,并且在某些位置可能发生不太有利的氨基酸。实际上,对最常观察到的氨基酸的突变通常会增加稳定性(共识效应)(Lehmann等,2000; Magliery,2015; Steipe等,1994)。但是,我们的方法本身并未实现共识。最重要的是,比对扫描消除了在自然多样性中罕见或从未见过的突变,而不是严格选择最常见的氨基酸。
接下来,我们应用Rosetta计算突变扫描(Whitehead等,2012),其中来自前一步骤的每个“允许的”突变单独建模在野生型结构的背景下,以及野生型之间的能量差异。类型和单点突变体计算(△△Gcalc)。通过单独计算每个突变的影响,而不是与其他突变相结合,我们将设计选择限制为可能对稳定性做出附加贡献的突变,而不是非加性和依赖于上下文的贡献,从而最大限度地降低误报风险。因此,我们将“潜在稳定”突变的空间定义为具有△△Gcal ≤ - 0.45 Rosetta能量单位(R.e.u。)的突变;我们选择这个截止而不是0 R.e.u.进一步降低虚假积极性的风险,如下文所述的系统评估所示。作为最后一步,我们使用Rosetta组合序列设计在潜在稳定突变的空间内找到最佳突变组合。此外,我们使用较低的△△Gcalc阈值在组合序列设计之前选择潜在的稳定突变,从而产生几种用于实验测试的设计(参见在线获得的数据S1)。
人乙酰胆碱酯酶(hAChE)中Gly416突变的选择说明了这两种滤膜(比对扫描和计算突变扫描)在修剪假阳性中的作用(图1A)。 位置416位于部分暴露的螺旋表面上,其中小而柔韧的氨基酸Gly可能使hAChE不稳定。 事实上,在AChE同源物的排列中,Gly很少见,而His是最常见的氨基酸。 然而,建模表明,在hAChE的特定背景下,His采用了紧张的侧链构象; 相比之下,由于其高螺旋倾向和与Tyr503的有利氢键结合,预计Gln是第三种最普遍的氨基酸,它是最稳定的。 因此,组合滤过器有利于Gln而不是His用于下游设计计算。
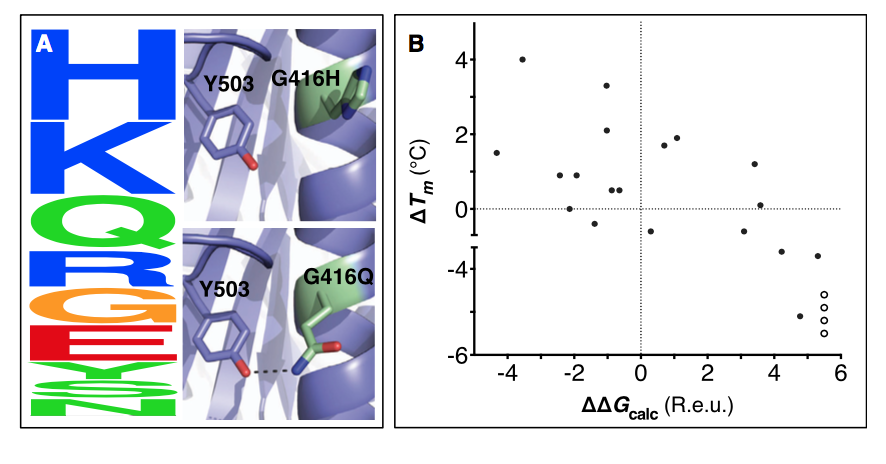
图1.通过同源序列分析和计算突变扫描消除潜在的不稳定突变
(A)(左)hAChE位置Gly416的Sequence logo。字母高度代表同源AChE序列比对中各自的氨基酸频率。位置416处的进化'‘允许的’序列空间(PSSM得分R0)包括所示的9个氨基酸。 (右)进化上有利的氨基酸His和Gln的突变的结构模型,其受Rosetta能量计算的青睐。 His侧链由于接近庞大的Tyr503芳环而变形,而Gln侧链松弛并与Tyr503形成有利的氢键(虚线)。 (B)计算突变扫描正确 鉴定酵母磷酸丙糖异构酶(TIM)中的所有去稳定突变和2/3稳定突变。使用共识设计(consensus design)来预测23种突变,这些突变将稳定酵母TIM并测量每种突变体相对于野生型(△Tm)的Tm差异。我们使用计算突变扫描来预测每个突变对稳定性的影响(△△Gcalc)。通过实验发现四个突变是高度有害的,导致没有功能性表达,因此Tm为 这些无法衡量(空心圆圈);这四种突变具有非常不利的△△Gcalc值(> 4.6 Rosetta能量单位; R.e.u。)。
为了系统地评估我们的过滤方法识别稳定突变的能力,我们将其输出与已发表的关于酶真菌内切葡聚糖酶Cel5A和酵母磷酸丙糖异构酶(TIM)中单点突变的稳定性影响的实验数据进行了比较(图1B;表1)(Sul- livan等,2012; Trudeau等,2014)。两个过滤器(PSSM和使用△△Gcalc≤-0.45 Reu作为截断值的Rosetta突变扫描)消除了所有严重不稳定突变(和所有去稳定突变的99.6%),并分别保留了Cel5A和TIM中实验验证的稳定突变的三分之一和三分之二。因此,对于这两种酶,我们的方法确定了大部分已知的稳定突变,同时几乎完全排除了假阳性。能量截止强调了最小化假阳性预测的重要性,即实验上不稳定但在计算上被认为是有利的突变;例如,在Cel5A的情况下,使用△△Gcalc<0 R.e.u,虽然截止值会正确地预测四种额外的稳定突变,但会引入另外八种不稳定突变。假阴性预测 - 即在实验中稳定但通过建模被认为是不稳定的突变 - 不能归因于单个建模误差(表S1)。具体而言,在Cel5A中的22个假阴性中,有8个被过滤,因为它们根据序列比对不太可能(PSSM得分<0),4个显示△△Gcalc在0.45到0 Reu范围内,从而略微忽略能量阈值。在剩余的10个假阴性中,有3个发生在核心位置,其中能量计算通常会对突变产生不利影响,其中2个是脯氨酸突变。能量函数和构想灵活度程序的改进可能在未来提高准确性,这里报告的分析为这些改进提供了基准。

三、结果
3.1 设计的hAChE具有近2,000倍的高细菌表达和完整的活性
我们将我们的设计策略应用于人乙酰胆碱酯酶(hAChE),这是一种60kDa的酶,通过快速水解神经递质乙酰胆碱(ACh)来终止胆碱能神经突触的突触传递(Sussman等,1991)。尽管AChE可能对有机磷酸盐神经毒剂和杀虫剂的检测和解毒有用,但目前hAChE是通过昂贵的程序生产的,包括从血液红细胞膜中纯化,或使用植物或乳腺表达系统。以前在细菌中表达hAChE的尝试导致极低水平的可溶性和活性蛋白质(Fischer等,1993)。 AChE的活性位点位于深峡谷的底部,渗透到酶的中途(20Å),沿着峡谷的突变可以将ACh水解速率降低多达1,000倍(Ordentlich等,1995) )。为了提高hAChE的稳定性和表达水平而不改变其活性,我们对新设计的hAChE的允许序列空间施加了进一步的限制:在所有Rosetta建模模拟中,氨基酸在8 Å 内的侧链构象̊可逆性抑制剂E2020跨越活性位点峡谷的全长(Cheung等,2012),必须保持原生hAChE结构(表S2)。后一种限制与上述两种滤波器相结合,大大减少了可用于设计的序列空间。 hAChE(一种550残基酶)的理论序列空间为10^750,即使对于高级建模算法也难以辨认。相比之下,减少的序列空间的大小是10^31个序列,相当于24个氨基酸肽的完整计算设计,这是20世纪90年代已经解决的挑战(Dahiyat和Mayo,1997)。我们注意到,对于任何给定的△△Gcalc截止值,减少的序列空间导致组合序列优化收敛到相同或几乎相同的序列;这种融合在计算设计中并不常见(Fleishman等,2011),是非专家可重复性和使用的先决条件。
考虑到AChE的大尺寸,我们使用不同的△△Gcalc阈值设计了五种替代方案,相对于hAChE具有17-67个突变(表S3;数据S2),并将它们进行实验测试。编码野生型hAChE的合成基因和五种设计针对细菌翻译效率进行了优化,融合到硫氧还蛋白的C末端,并在大肠杆菌ShuffleT7express细胞中表达以促进二硫键形成。在来自细菌裂解物的上清液级分的SDS-PAGE凝胶中,AChE条带与其他条带重叠,排除了视觉定量。然而,由于发现设计的变体的特异性活性与野生型(表2)几乎相同,我们可以通过测量粗裂解物中的AChE活性来量化可溶性酶和活性酶的相对水平。与hAChE相比,五种设计显示R100倍高的裂解物ACh水解速率,最佳设计dAChE4(图2A; 51种突变),显示出高达近2,000倍的速率(图2B)。此外,由于其极低的可溶性表达,hAChE无法从粗细胞裂解物中纯化,而设计的变体易于纯化至同源性,标准摇瓶中的dAChE4产量接近每升细菌培养物2 mg蛋白质(表S4)。
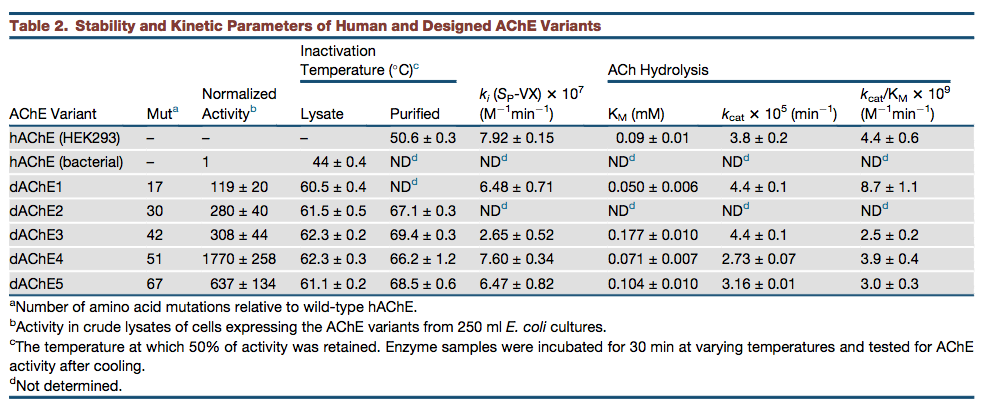
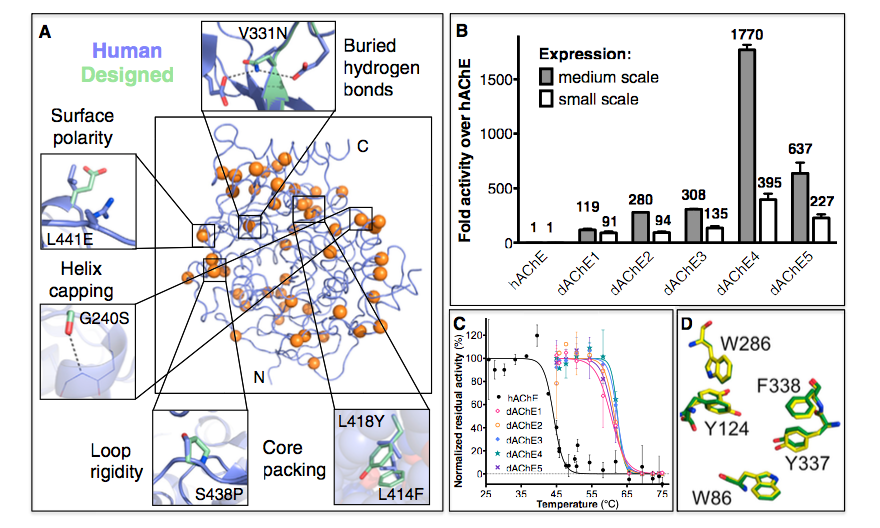
图2.稳定的hAChE变体的设计及其在细菌中的功能表达 (A)设计变型dAChE4中稳定化的结构基础。野生型hAChE以蓝色显示,51个突变位置分布在整个dAChE4中,由橙色球体表示。缩略图突出显示所选突变的稳定效应。 (B)设计的AChEs的细菌裂解物活性水平归一化为hAChE活性。粗裂解物来自250ml烧瓶(中等规模)或在96孔板(小规模)中生长的0.5ml大肠杆菌培养物。设计变体中较高的活性水平反映了较高水平的可溶性功能酶。 (C)与hAChE(黑色)相比,设计的AChE变体(彩色线)显示出更高的抗热失活性。在细菌裂解物中测量在不同温度下孵育后的残余活性,并将其标准化为未处理的裂解物中的活性。 (D)与hAChE(PDB:4EY4,绿色)相比,在dAChE4(PDB:5HQ3,黄色)的晶体结构中催化三联体附近的关键残基排列的亚-Å埃准确度。
设计的AChE突变分散在整个酶中,显示出稳定突变的典型特征,包括改善的核心堆积,更高的骨架刚性和增加的表面极性(图2A)。 与设计策略和较高水平的可溶性和功能性酶一致,我们观察到相对于大肠杆菌表达的hAChE(图2C)和在哺乳动物细胞中表达的hAChE,对高达20℃的热灭活的抗性增加(表2)。 设计水解ACh的速率在hAChE速率的2倍范围内,并且显示出与hAChE几乎相同的神经毒剂VX的失活速率常数(观察到dAChE3的最大偏差,表现为 灭活率降低2.5倍;表2)。
上述对设计的和野生型AChEs的几乎相同的活性谱的观察表明,设计的酶的活性位点基本上与hAChE的相同。为了验证这一点,我们对dAChE4进行了结晶试验,该设计表现出最高的细菌表达产量。我们注意到,与我们研究的各种天然AChEs相比,大型晶体已经在几天内形成并且更可重复。 dAChE4的结构以2.6Å分辨率求解,因此据我们所知,产生在原核生物中表达的AChE的第一个结构(表S5)。 dAChE4的结构与野生型hAChE的结构非常相似,与Ca原子相比具有0.7Å的均方根偏差(rmsd)。催化峡谷的残留物排列得特别好,全原子rmsd仅为0.125Å(图2D)。因此,尽管相对于野生型有51个突变,细菌表达水平增加了2,000倍,耐热性提高了20多克,但dAChE4在其活性位点上几乎不受hAChE的影响(酶的其他部分的差异显示在图S1和在线 http://proteopedia.org/w/Journal:Molecular_Cell:1 )。因此,dAChE4可用于未来针对AChE活性位点的抑制剂的结构研究。
3.2 其他设计的酶显示高稳定性和/或可溶性表达
我们将我们的算法应用于另外两种人类酶 his-tone deacylase SIRT6,人类DNA甲基转移酶Dnmt3a和一种称为PTE的细菌磷酸三酯酶。作为已知的唯一已经进化为降解有机磷酸盐(OPs)的酶,PTE具有相当大的生物技术潜力,可用于去除包括神经毒剂在内的OP的去污和解毒作用。然而,PTE以大于实际应用的速度解毒大多数神经毒剂。野生型PTE之前被设计用于更高表达(Roodveldt和Tawfik,2005)和对各种OP的水解速率(Big-ley等,2015; Cherny等,2013)。然而,功能改变突变的引入使酶不稳定,这通常是实验室进化酶的情况(Tokuriki等,2008);因此,稳定是进一步工程的先决条件(Bloom等,2006)。我们使用不同的△△Gcalc阈值设计了三种替代方案,编码相对于野生型PTE的9-28个突变(表S3),并进行实验测试。编码三种设计的合成基因和PTE-S5,一种编码三种突变的已发表变体,与野生型相比显示高20倍的表达水平(Rood-veldt和Tawfik,2005),针对细菌翻译效率进行了优化,融合至麦芽糖结合蛋白,并在大肠杆菌GG48细胞中表达。与PTE-S5相比,所有三种设计都显示出增加的可溶性功能酶水平,其已经显示出比野生型更高的表达水平(表3)。三种设计中的两种相对于PTE-S5显示出高于10℃的热灭活耐受性,对OP底物对氧磷的活性没有显着变化(表3;图S2A)。稳定设计的另一个值得注意的结果是增加了金属亲和力。 PTE是含有两种活性位点金属的金属酶,通常为Zn + 2(Benning等,2001)。用于高表达的野生型PTE的定向进化(PTE-S5)导致金属亲和力的显着降低 - 这是在不能补充Zn + 2的条件下应用的主要实际缺点。设计的变体dPTE2含有19个突变并表现出最高的热灭活耐受性,与PTE-S5相比,金属亲和力显着增加,接近野生型PTE的亲和力(图3A和S2B;表3))。
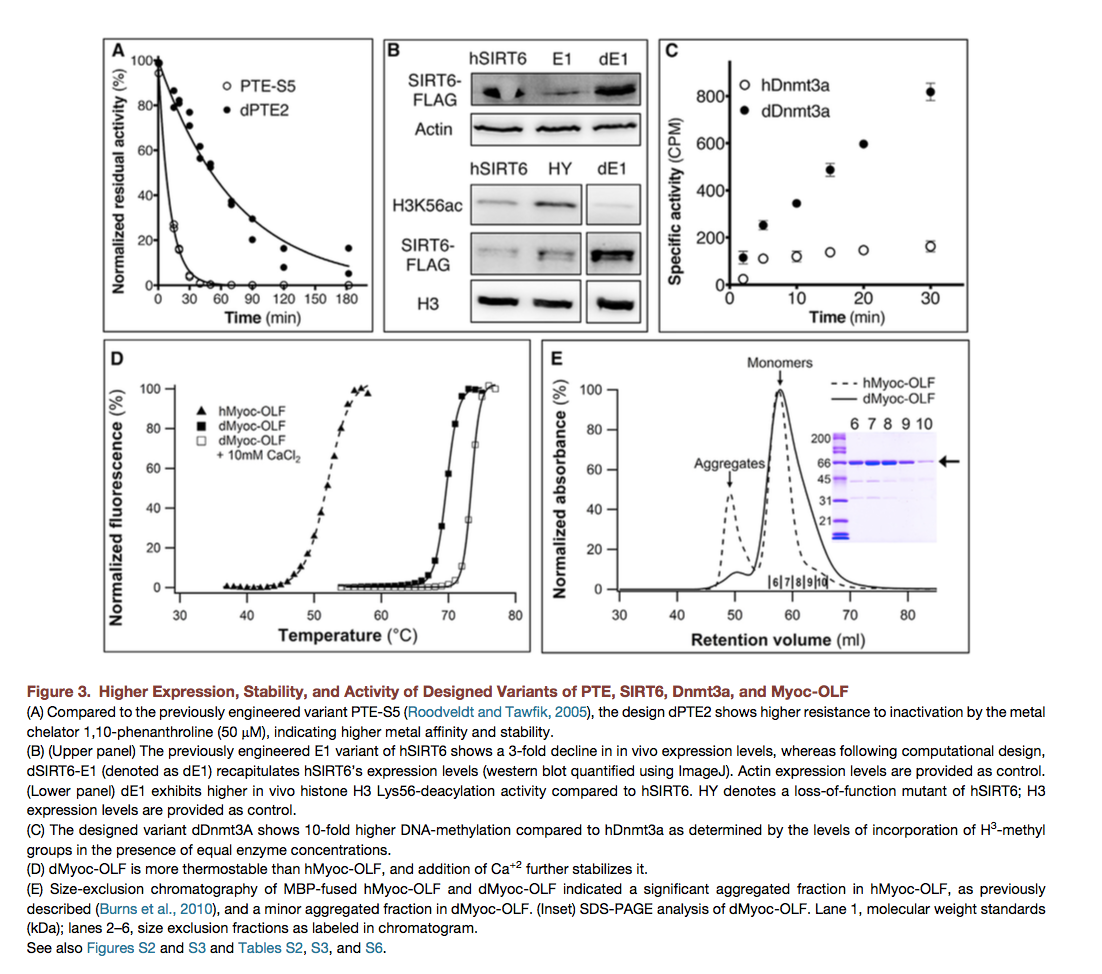
SIRT6是一种ADP-核糖基酶和NAD +依赖性脱酰酶,可去除酰化赖氨酸中的酰基,从而调节几种必需的细胞过程(Kugel和Mosto-slavsky,2014)。人SIRT6(hSIRT6)的低细菌表达水平及其相对于SIRT1的弱脱酰活性限制了其研究。设计的变体(dSIRT6)含有相对于hSIRT6的11个突变(表S2和S3)。将其在大肠杆菌中表达并纯化至同质,每升培养物20mg,相对于hSIRT6增加5倍,其脱酰酶活性比hSIRT6高60%(图S3A和S3B)。我们随后将设计突变整合到一个名为E1的工程化hSIRT6变体的背景中,该变体包含三个相对于hSIRT6的突变(我们未发表的数据)。与hSIRT6相比,E1表现出增加的脱酰活性,但在人细胞系中表达水平降低3倍。相比之下,设计的变体dE1重现了人类细胞系中hSIRT6的表达水平,同时保持了高的脱酰活性(图3B)。考虑到SIRT6过表达对小鼠寿命的有益影响(Kanfi等,2012),我们设计的突变体可用于建立SIRT6的去酰化率与其生理功能之间的相关性。
在人Dnmt3a(hDnmt3a)的情况下,可通过大肠杆菌表达获得人酶的可溶性和活性级分,但酶活性非常低。 我们测试了一种设计dDnmt3a,其含有相对于hDnmt3a的14个突变(表S2和S3)。 与所有其他设计相反,设计的dDnmt3a在大肠杆菌中显示出比hDnmt3a显着更低的表达水平(图S3C和S3D)。 尽管如此,它的特异性活性高出近10倍(图3C),表明hDnmt3a制剂含有大量可溶性但无活性或活性差的酶。 因此,尽管Dnmt3a未能满足我们的设计方法的成功标准(更高的稳定性和可溶性表达),但它显着提高了蛋白质的功能产量。
3.3 用于蛋白质稳定化的Web服务器
受到上述一致积极结果的鼓舞,我们将算法实施为网络服务器,称为Protein Repair One Stop Shop(PROSS,http:// pross.weizmann.ac.il)。 请求帮助解决关于人肌球蛋白OLF结构域的关键表达和稳定性问题(hMyoc-OLF),两位作者(S.E.H和R.L.L)被授予无监督访问Web服务器的权限。 OLF结构域存在于多细胞生物的细胞外蛋白中。 许多OLF结构域与人类疾病有关,但大多数OLF结构域的功能仍然难以捉摸。 研究最多的OLF结构域是hMyoc-OLF,其中超过100个非同义突变与遗传性开角型青光眼有关。 患者中记录的突变导致形成细胞毒性聚集的不稳定肌球蛋白(Burns等,2010; Yam等,2007)。S.E.H. 和R.L.L. 因此认为,赋予增强稳定性的突变可能会降低hMyoc-OLF在生理温度下错误折叠的倾向。
三个hMyoc-OLF结构(PDB:4WXQ,4WXS和4WXU)被提交给PROSS,每个结构产生七个设计,每个结构有5-25个突变。 从所有建议的点突变,S.E.H。和R.L.L. 手动衍生的一个变体(dMyoc-OLF)包含21个突变。 虽然hMyoc-OLF结合Ca + 2(Donegan等,2012),但Ca + 2结合的生理作用仍然未知; 然而,Ca + 2结合位置保持不变,反映了作用于它们的高序列和结构限制。 此外,没有任何设计突变与人类疾病有关,支持了与疾病相关的突变不稳定的观念。
dMyoc-OLF和hMyoc-OLF在与麦芽糖结合蛋白(MBP-OLF)融合的大肠杆菌中表达。 设计的变体比hMyoc-OLF产量高一个数量级(表S6)。 此外,虽然纯化的hMyoc-OLF包含可溶性MBP-OLF聚集体和正确折叠的单体的混合物(Burns等人,2010),但设计的变体几乎是单体的,并且还显示出 熔点温度(Tm)高于hMyoc-OLF(图3D和3E)。 这是目前为Myoc-OLF测量的最高Tm,几乎与缺乏Ca +2结合位点的gliomedin OLF结构域相同(Hill等,2015)。 尽管如此,如存在Ca + 2时Tm增加所示(图3D;表S6),在dMyoc-OLF中仍然存在Ca + 2结合。 因此,设计的变体dMyoc-OLF是详细的OLF结构 - 功能分析的起点
四、讨论
大多数现有的稳定性设计方法关注蛋白质稳定性的一个或另一个要素。例如:
- 一些方法引入二硫化物或脯氨酸,
- 而另一些方法优化核心包装(Borgo和Havranek,2012; Korkegian等,2005)
- 并增加表面极性(Magliery,2015)和电荷(Lawrence等,2007) 。
相反,我们的设计算法选择优化蛋白质计算能量的所有氨基酸突变,受限于同源序列推断的限制。因此,它可以生成设计,改善一系列与稳定性相关的分子参数(表4),同时保留功能。此外,我们发现,在这里研究的各种蛋白质中,该算法实施的解决方案似乎选择性地解决了每种蛋白质的特定缺陷。例如,设计dAChE4的51个突变中的17个影响核心位置,与之前针对蛋白质核心的设计研究相比,这是非常大的数量(Borgo和Havranek,2012; Korkegian等,2005)。实际上,天然AChE结构包含异常大的核心腔(Koellner等,2000)。相比之下,在设计dPTE2中,仅实施了两个核心突变,净负电荷增加了13个单位,将等电点(pI)从野生型的近中性值(6.7)转移到5.0。 ;实际上,近中性pI通常与溶解性差有关,并且其他人已经使用表面“增压”来改善热稳定性(Lawrence等,2007)。然而,稳定性考虑不是以功能约束为代价的。例如,在Dnmt3a中,正的净电荷和碱性pI完全保守,如DNA结合蛋白所预期的那样。我们得出结论,进化限制的组合,单独有助于计算稳定性的突变的选择,以及这些突变空间内的组合序列优化可以解决各种蛋白质中的广泛稳定性缺陷而不影响其原始分子功能。
这里讨论的案例研究代表了属于不同折叠家族(TIM barrel, b-propel- ler, and α/β hydrolase fold)并显示不同活性的多种蛋白质。在每种情况下,先前使用传统方法工程化的野生型蛋白质和变体都具有低稳定性,如通过对高温和功能修饰突变的不耐受或通过低溶解度,异源表达,辅因子亲和力或特异性所致。只考虑系统发育约束并优化天然状态能量的单一全自动方法能够在不牺牲功能的情况下解决这些不同的挑战,这表明这些问题的根本原因是天然状态的边际稳定性。此外,稳定性设计方法有可能优化用作从头粘合剂或酶设计的支架的蛋白质,其中低稳定性和表达水平受到限制(Fleishman等,2011; Khersonsky等,2011) 。
四、限制
稳定性设计算法需要几十种独特的序列同源物和靶蛋白的原子结构。 随着序列数据库的迅速发展,第一个要求可能会被满足,只有少数例外; 然而,第二种可能是一种障碍,特别是因为低蛋白质产量和稳定性会对结构测定产生负面影响。 为了扩展到实验确定的结构之外,可以使用同源模型,并且它们是否为原子稳定性设计计算提供足够的准确度还有待观察。
五、实验过程
5.1 计算程序–系统发育序列约束
对于每个查询序列,使用BLASTP算法(Altschul等,1990)在非冗余(nr)数据库上收集同源序列,最大命中数设置为500,e值为10^-4.所有其他参数均为 设置为默认值。 如果它们覆盖小于查询序列的60%或者它们与查询序列的序列同一性低于30%(由于同源的低多样性在> 30%序列同一性下,PTE为28%),则排除命中。 剩下的序列使用cd-hit进行聚类(Li和Godzik,2006),聚类阈值为90%,默认参数。
MUSCLE(Edgar,2004)与默认参数一起用于从聚类序列中导出多重序列比对(MSA)。查询和任何同源物之间的比对中的空位通常发生在环区域内,并且可以反映局部骨架构象的差异。 为了减少比对不确定性,我们检测了查询蛋白质结构中的二级结构元素(使用DSSP [Kabsch和Sander,1983])并消除了包含相对于查询的任何两个二级结构元素之间的间隙的同源物中的子序列。 。 实际上,对于每个环区域,该过程产生特异性比对,其仅包含相对于查询没有插入或缺失的同源序列。由于在我们设计AChE和Dnmt3a时尚未实施稳定性设计算法的这一特征,因此这两种蛋白质设计为默认MUSCLE比对,包含来自环区域中所有同源物的信息。 这一步不是太理解?
给定每个查询序列的比对,我们使用PSI-BLAST算法计算了位置特定的评分矩阵(PSSM)(Altschul等,2009)。 PSSM表示在查询中观察每个位置的20个氨基酸中的每一个的对数概率。 非负PSSM评分被认为可能在进化中发生并定义允许突变的空间。
5.2 基于结构的约束
为了防止活性丧失,在活性位点(hAChE和Dnmt3a)中观察到小分子配体的8Å内的残基,或者在5 Å的金属辅助因子(PTE)中,DNA链(Dnmt3a)和蛋白质配体(SIRT6)在所有Rosetta模拟中保持固定。 在同型寡聚体结构(PTE和AChE)中,寡聚体界面的5A’内的残基保持固定。对于PTE和SIRT6,一些活性位点限制是使用与另一个配体结合结构的比对来定义的,即4NP7中DPE的DPJ的8Å 内的残基,以及3ZG6中链F的5Å 内的残基。 SIRT6末端(如果末端不与蛋白质的其他部分发生相互作用)不允许发生突变。对于hSIRT6,发现在先前的实验室进化实验中增加活性的四个残基在模拟之前被建模,并且不允许突变(参见表S2)。在PROSS网络服务器中,活动站点约束遵循上面定义的相同阈值,并且用户可以通过交互式HTML表单指定在模拟期间要保持固定的任何其他残留。
5.3 Rosetta结构建模与设计
在整个模拟过程中交替出现了两个能量函数:
- 全原子’hard-repulsive’ Rosetta能量函数(talaris2014)(O’Meara等,2015),由范德瓦尔斯,氢键,隐性溶剂化, 具有距离依赖的介电系数的库仑静电,
- 以及该能量函数(soft-rep)的“soft-repulsive”版本,其中范德华重叠和残余构象应变被降低。两个能量函数增加了谐波主干坐标限制设置为在PDB结构中观察到的坐标,和 术语有利于PSSM评分较高的突变。
我们通过四次迭代的侧链填充(活性位点残基除外)和侧链和骨架最小化来改进每个PDB结构,从而节省了最小能量结构。对于hAChE,这里测试的最大蛋白质,细化计算平均每条轨迹2.5小时(数据S1)。
使用Rosetta中的FilterScan mover将计算突变扫描应用于精制结构(Whitehead等人,2012)。在每个位置,每个允许的突变(即,与PSSM得分≥0的每个氨基酸同一性)在精制结构的背景下单独建模。模拟突变的8Å内的蛋白质侧链被重新包装,并且使用侧链和约束骨架最小化来适应突变。使用ta-laris2014(O’Meara等,2015)计算精制结构和单点突变体的优化构型之间的能量差异。七个能量阈值用于定义不同的突变空间(-0.45,-0.75,-1.0,-1.25,-1.5,-1.8和-2.0 R.e.u.)。变异扫描是计算中最耗时的步骤,使用标准CPU(数据S1)每个位置需要30秒到6分钟(hAChE平均每个位置1分钟)。该程序可以并行化我们 - 在PROSS网络服务器中实现的计算机集群。
对于Dnmt3a,从允许的序列空间中消除了9个突变,因为它们的Rosetta backbone-omega能量显示出异常高且意外的变化,这可能是由于输入PDB结构的结晶分辨率差(表S2)。
对于潜在稳定突变的每个序列空间,实施了组合序列优化。从精细结构开始,我们对PDB文件中的坐标施加了坐标约束,并实现了序列设计,侧链和骨架最小化的四次迭代,同时交替soft and hard repulsive potentials。对于AChE,每个设计轨迹平均花费2.5小时(数据S1)。
5.4 设计模型分析
将设计的变体的序列和结构特征与野生型蛋白质的序列和结构进行比较。
使用Rosetta Features Reporter套件(Leaver-Fay等人,2013; O’Meara等人,2015)评估盐桥和氢键数量的差异,小于5.5 Å 的盐桥相互作用定义为两个反电荷。
埋藏残留物被定义为分别在10Å 和12Å 内具有> 22和> 75个相邻非氢原子的残基。
使用ExPASy网络服务器( http://web.expasy.org/compute_pi )计算蛋白质等电点(pI)。
有关实验程序,请参阅在线补充实验程序。 对于DNA和蛋白质序列,请参见数据S2。
讨论
这个△△Gcalc这么有用,是怎么来的呢?
参考资料
- Goldenzweig, A., Goldsmith, M., Hill, S. E., Gertman, O., Laurino, P., Ashani, Y., … Fleishman, S. J. (2016). Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Molecular Cell, 63(2), 337–346.
