【4.4.1】蛋白质结构预测和设计的进展
名词解释:
-
蛋白质能量函数(Protein energy functions): 对应于确定蛋白质结构和相互作用的分子力数学模型的功能。 能量函数的选择定义了从结构到能量值的映射,称为能量景观( energy landscap),它可以指导结构预测和设计模拟。 典型的蛋白质能量函数是多个项的线性组合,每个项都捕获不同的能量贡献(范德华相互作用,静电,去溶剂化),而这些项的权重和原子参数是通过参数化程序来选择的,这些参数化程序力求优化之间的一致性, 从能量函数预测的数量以及从实验或小型化学系统上的量子化学计算得出的相应值
-
范德华相互作用 (Van der Waals interactions): 原子间或分子间的相互作用较弱(比共价键或离子键弱得多)且相对较短。
-
旋转器 (Rotamers):氨基酸侧链经常采用的一组离散构象。
-
自由程度 (Degrees of freedom): 系统中的自由参数决定了它的结构,从而决定了它的能量。 它们可以是连续的,例如实值主干扭转角或原子位置,也可以是离散的(仅允许有限数量的替代方法)。 由于强烈的扭转偏好,可以使用离散的一组旋转异构体成功建模侧链构象,并通过对结构数据库的分析加以识别。
-
披角(Phi angle): 扭转角(或二面角),描述围绕连接多肽链中氨基酸的骨架氮和骨架Cα碳原子的键的旋转。 它是每个氨基酸残基两个主要的自由度之一(连同psi角),蛋白质用来采用其他构象。
-
Psi angle: 扭转角(或二面角),描述围绕连接多肽链中氨基酸的骨架Cα碳和骨架羰基碳的键的旋转。 它是每个氨基酸残基两个主要的自由度之一(连同phi角),蛋白质用来采用其他构象。
-
Chi1 angle: 氨基酸侧链的扭转角(或二面角)是根据键与蛋白质主链化学连通性的接近程度来编号的。 Chi1是指围绕最接近骨架的键旋转,chi2是下一个最接近的位置,依此类推。 一些氨基酸(例如丙氨酸和甘氨酸)没有可旋转的键或扭转角,而其他氨基酸(例如赖氨酸和精氨酸)最多有四个。
-
消除死角(Dead-end elimination): 一种用于侧链旋转异构体优化的算法,该算法通过消除与采用最低能量的序列不兼容的旋转异构体而起作用。
-
平均场优化(Mean-field optimization): 一种用于设计序列的协议,该协议为观察蛋白质中每个序列位置的每个氨基酸分配一个概率,并根据分配的概率计算蛋白质的平均(平均场)能量。 然后调整概率以降低蛋白质的平均场能。
-
模拟退火(Simulated annealing): 一种概率低的方法,用于根据序列发生改变时蛋白质能量的计算变化和建模系统的温度来确定接受或拒绝序列变化的低能序列。 如果变化降低了蛋白质的能量,它会被自动接受; 如果它提高了系统的能量,则被接受的可能性很大,这取决于能量增加了多少(能量增加的可能性较小,不太可能被接受)和当前温度(在较高温度下,则更有可能 接受会增加系统能量的更改)。 随着模拟的进行,温度会降低,以识别低能序列。
-
遗传算法(Genetic algorithms): 一种序列优化协议,通过应用基于能量的选择轮次来重复修改序列总体。 通过将序列建模为所需的蛋白质构象,可以计算出序列的能量。 能量较低的序列更有可能发展到下一代。 在每一轮选择之前,将先前的获胜序列彼此重组,并在序列中掺入少量突变,以寻找能量较低的序列。
-
多状态设计算法(Multi-state design algorithms): 一种设计同时满足多个约束的序列的方法。 例如,此类算法可用于查找被预测与配体x结合但不与配体Y结合的蛋白质序列。或者,可将它们重新用于查找同时擅长结合配体x和配体Y的序列。
-
酵母表面展示(Yeast surface display): 一种探索大型蛋白质序列文库(多达数千万个)与另一个分子结合的实验方法。 在最终的酵母文库中,每个酵母细胞都包含蛋白质文库一个成员的DNA,该蛋白质表达为融合蛋白,将蛋白质呈现在细胞外。 将细胞与已用荧光染料标记的靶分子混合,然后使用荧光激活的细胞分选法鉴定包含结合靶蛋白的设计蛋白的细胞。 DNA测序用于鉴定通过选择的设计。
-
BCL-2蛋白家族(BCL-2 protein family): 彼此相互作用以诱导或抑制细胞凋亡的结构相关蛋白家族。
-
DNA折纸(DNA origami): 一个描述使用DNA相互作用的高序列特异性来设计可折叠成复杂且可预测的二维和三维形状的DNA序列的方法的术语。
-
主要组织相容性复合体(Major histocompatibility complexes,MHC): 一组细胞表面蛋白,可与外源病原体的抗原结合,并呈递它们以被免疫系统中的其他蛋白和细胞识别。 它们是获得性免疫系统的关键组成部分。
摘要:
几十年来,由于其固有的科学兴趣以及从基因组解释到蛋白质功能的强大蛋白质结构预测算法的许多潜在应用,从氨基酸序列预测蛋白质三维结构一直是计算生物物理学中的一大难题。最近,反问题(inverse problem)-设计将折叠成指定的三维结构的氨基酸序列-作为合理地工程化具有生物技术和医学功能的蛋白质的潜在途径,引起了越来越多的关注。在过去的十年中,用于预测和设计蛋白质结构的方法取得了巨大的进步。计算能力的提高以及蛋白质序列和结构数据库的快速增长,推动了新的数据密集型和计算要求高的结构预测方法的发展。设计蛋白质折叠和蛋白质-蛋白质界面的新算法已用于工程化新型高阶装配,并从头开始设计具有新特性或增强特性的荧光蛋白,以及具有治疗潜力的信号蛋白。在本综述中,我们描述了蛋白质结构预测和设计的当前方法,并重点介绍了它们已启用的成功应用程序。
天然进化的蛋白质所具有的惊人的分子功能多样性,是由于其微调的三维结构而得以实现的,而三维结构又由其遗传编码的氨基酸序列决定。因此,对氨基酸序列与蛋白质结构之间关系的预测性理解将开辟新途径,既可用于根据基因组序列数据预测功能,也可通过设计具有特定结构的氨基酸序列来合理地设计新型蛋白质功能。在过去的十年中,我们预测和设计蛋白质三维结构的能力得到了显着提高,这对医学和我们对生物学的理解具有潜在的深远影响。已经开发出了新的机器学习算法,可以分析蛋白质家族中相关突变的模式,从而仅从序列信息中预测结构上相互作用的残基。改进的蛋白质能量功能首次使从近似的结构预测模型开始,并通过能量引导的精制过程使其更接近实验确定的结构成为可能。蛋白质构象采样和序列优化方面的进展已允许设计新的蛋白质结构和复合物,其中一些具有治疗前景。
蛋白质结构预测和设计的这些进步,由于技术上的突破以及生物学数据库的迅速发展而得到了推动。蛋白质建模算法(方框1)在计算上既需要开发也要应用。研究人员可利用的计算能力的迅速提高(包括基于CPU的计算能力,以及越来越多的基于GPU的计算能力)都有助于对新算法进行快速基准测试,并使它们能够应用于更大的分子和分子组装体。同时,随着基因组和宏基因组测序工作的扩大,下一代测序已大大促进了蛋白质序列数据库的发展。软件和自动化技术的进步加快了实验结构确定的步伐,加快了实验确定的蛋白质结构数据库(蛋白质数据库(PDB))的增长,该数据库现已包含近150,000个大分子结构。寻求利用这些扩展序列和结构数据库的蛋白质建模人员,现在正在采用彻底改变图像处理和语音识别的深度学习算法。

框1:浏览蛋白质能量图景 。蛋白质构象能图是复杂的高维表面,具有许多局部最小值。在这些景观中导航以定位低能耗盆地以进行预测和设计需要有效的采样方法和准确的能量函数。在基于梯度的优化方法中(参见左上图),计算能量函数相对于柔性自由度的导数(例如原子坐标或主链扭转角),以便沿能量下降最快。基于梯度的优化可以有效地找到能源格局中最接近的局部最小值,但通常不会找到全局最小值。蒙特卡洛采样方法采用随机选择的构象运动和偶然的上坡步骤来逃避局部最小值(参见下图)。在蒙特卡洛大都会(Metropolis Monte Carlo44)中,根据能量变化接受(绿色箭头)或拒绝(红色箭头)采样运动:降低能量的下坡运动以概率1被接受,而上坡运动(虚线箭头)被接受为随能量变化呈指数下降的概率P。用于蒙特卡洛模拟的移动集的示例包括片段替换移动,其中,当前构象中的连续骨架段被片段库中的替代构象替换,以及侧链旋转异构体替代。蒙特卡洛采样的一种流行替代方法是分子动力学模拟(参见右上图),其中构象采样是由牛顿运动定律决定的,该定律适用于分子系统的势能函数。给定一组初始的原子位置和速度,作用在每个原子上的力通过采取势能的梯度来计算,然后从牛顿第二定律(F = ma)得出加速度。在时间上迈出了非常小的一步(通常是几飞秒的数量级),并且新的位置和速度是根据时间步长的大小以及旧位置,速度和加速度来计算的。凭借精确的能量函数和足够小的时间步长,长期的分子动力学模拟提供了广泛的能量分布样本,并且给出了各个分子如何随时间演化的逼真的图像。基于分子动力学的建模方法面临的挑战是,必须执行数百万至数万亿个时间步长才能达到生物学相关的时标,这需要高性能的软件,在某些情况下甚至需要专用的超级计算机
在这篇评论中,我们重点介绍了这些技术进步带来的近期突破。 我们描述了蛋白质结构的预测和设计的当前方法,主要侧重于无需模板的方法,该方法不需要将实验确定的结构作为模板。 将讨论这些建模方法的优缺点以及它们当前和潜在的应用。 最后,我们评论这些发展对生物学和医学领域的更广泛的实践意义。
一、蛋白质折叠力 Protein-folding forces
蛋白质具有自发折叠成精确确定的三维结构的卓越能力。重新折叠实验已经确定,指定蛋白质折叠构象(其天然状态)所需的信息完全包含在其线性氨基酸序列中。根据安芬森(Anfinsen)的热力学假设,此信息以多肽的能量构图形式编码:天然状态是自由能最低的状态。该假设为蛋白质结构预测的通用方法奠定了基础,该方法将替代构象的采样与评分相结合,以按能量对它们进行排序,并确定最低能量状态。 Cyrus Levinthal首先将这种能量指导方法成功的主要障碍是在生物时间尺度上将蛋白质折叠的概念性障碍,但其潜在的构象空间很大:即使假设每个氨基酸只有有限的离散集合在可能的骨架状态中,必须搜索的构象空间的总大小随链长成指数增长,并且很快就变成了天文数字。解决这一难题的方法是认识到,不必为了确定原始状态而探索整个构象空间:能源格局并不是一个带有单个“洞”的平坦“高尔夫球场”;相反,方向性提示将整体漏斗形状赋予景观,并引导采样指near-native的构象(图1a)。这些方向性提示可能来自于序列-局部残基相互作用,使链的短链偏向形成特定的二级结构,也可能源自甚至在达到全球天然折叠之前就可能形成的有利的,长期的,非局部的包装相互作用。

图1 |蛋白质折叠态势和能量( Protein-folding landscapes and energies)。一个| “高尔夫球场”和“漏斗”形的能源格局的简化二维表示。确定左侧景观中的native最低能量(‘N’)需要详尽的探索,而从大多数起点进行简单的下坡搜索将在右侧景观中找到native状态。 b |区分蛋白质天然状态的能量特征包括:疏水模式(此处显示为小蛋白质泛素的剖面图),在蛋白质核心中埋有非极性侧链;骨架和侧链氢键(氢键显示为绿色虚线);紧密的侧链堆积(在穿过蛋白质核心的切片中可见);以及受限的主链和侧链扭转角分布(在氨基酸的异亮氨酸的主链高度集中的二维概率分布中-phi角与psi角以及侧链-chi1角与chi2角-扭转角)。 c |蛋白质能量学的计算模型提供了速度和准确性之间的权衡。粗粒度模型(Coarse-grained models )具有高效的计算能力,可以有效地平滑能源格局,允许进行大规模采样;但是,它们还会引入不正确的信息,例如错误的极小值(例如,该部分中native极小值左侧的蓝色盆地,用箭头突出显示)。高分辨率,原子详细的能量函数更精确,但评估速度也较慢,并且对结构细节敏感,这会在景观中引入凹凸不平(许多局部最小值),使它们难以有效导航。
人们认为推动水溶性球状蛋白质折叠的驱动力是将疏水性氨基酸侧链从水中掩埋。折叠与结构性熵的损失相反,结构性熵的丧失伴随着柔性多肽链塌陷成确定的3D构象。蛋白质核心中非极性侧链的紧密堆积增强了范德华力的相互作用,并消除了在熵方面不利的内腔(图1b)。此外,这种拼图游戏般的堆积是在实现强主干和侧链扭转优先权的同时,限制了观察到的扭转角分布(图1b中的下部面板),从而有效地降低了侧链的柔韧性。每个位置都有一组离散的旋转器(rotamers)。蛋白质内的氢键和盐桥在很大程度上补偿了与水相互作用的损失,因为极性基团在折叠过程中被掩埋,因此这些相互作用对天然状态的稳定性的贡献小于对天然状态的特异性的贡献(也就是说,它们有助于区分native状态与其他紧凑状态)。尽管可以从低分辨率结构模型中检测到疏水性掩埋和主链氢键结合,但紧密的核堆积以及不存在区分天然状态的掩埋的,不满意的极性基团,都需要对侧链自由度进行显式建模。结果,用于结构预测和设计的分子建模方法通常采用多个级别的分辨率:使用具有计算效率的粗粒度能量函数执行大规模构象采样,该函数捕获疏水埋葬,形成二级结构并避免原子重叠;最终的蛋白质模型选择和完善需要使用更耗时的高分辨率原子能函数对氨基酸侧链进行explicit建模(图1c)。
二、蛋白质结构预测
有两种通用方法可以预测目标蛋白质(“靶标”)的结构:
- 基于模板的建模,其中先前确定的相关蛋白结构用于模拟靶标的未知结构;
- 无模板建模,它不依赖于PDB中结构的全局相似性,因此可以应用于具有新折叠的蛋白质。
从历史上看,这两种方法所采用的方法是截然不同的,基于模板的建模专注于对已知结构的相关蛋白质的检测和比对,而依赖大规模构象采样的无模板建模以及基于物理的能量函数的应用。但是,近来,这些方法之间的界线已开始模糊,因为基于模板的方法已结合了能量指导的模型改进,且无模板方法采用了机器学习和基于片段的采样方法来利用结构数据库中的信息(尽管基于模板的方法仍然可以使目标序列的准确性与PDB中的条目相似,但仍保持较高的准确性)。在这里,我们简要介绍了基于模板的建模方法,然后转向无模板建模,并描述了该领域的最新发展。
2.1 基于模板的建模
基于标准模板的建模步骤包括:
- 选择合适的结构模板;
- 靶序列与模板结构的比对;
- 分子模型来解释靶标-模板比对中存在的突变,插入和缺失。
可以通过使用单序列搜索方法(例如BLAST)扫描PDB序列来检测紧密相关的模板。为了检测更远距离相关的模板,可以使用由多序列比对构建的靶序列图谱,通过图谱-图谱(profile–profile)比较来扫描序列图谱数据库中已知结构的蛋白质,或者可以将其与结构模板库进行匹配评估序列结构兼容性。模板选择方法返回初始的目标模板对齐方式,该对齐方式可以手动建立,通常在模型构建后以迭代方式进行调整。给定与模板的比对,可以通过仅在突变的位置进行侧链优化并通过围绕插入和缺失重建骨架,来使用已建立的工具来快速构建靶序列的分子模型。对于仅与已知结构的蛋白质有远距离关联的靶蛋白序列,可能需要更复杂的方法,这些方法依赖于多个模板并执行具有侵略性的骨架构象采样。与可用的晶体结构一起,基于模板的建模方法可以为大约三分之二的已知蛋白质家族提供结构信息。
2.2 免模板建模 Template-free modelling
无模板建模方法可以应用于蛋白质,而与PDB中的蛋白质没有全局结构相似性。 缺少结构模板,这些方法需要用于生成候选模型的构象采样策略,以及用于选择类似天然构象的排序标准。 没有模板的结构预测过程(图2):
- 通常从构建目标蛋白和相关序列的多序列比对开始。
- 然后,将靶标及其同源物的序列用于预测局部结构特征,例如二级结构和主链扭转角,以及非局部特征,例如残基与残基的接触或整个多肽链上的残基间距离。
这些预测特征可指导构建目标蛋白质结构的3D模型的过程,然后对其进行精炼,排名和比较,以选择最终预测。

图2 |无模板结构预测中的关键步骤:
- 目标蛋白及其序列同源物之间的准确多序列比对,包含有关同源序列之间氨基酸变化的有价值信息,包括在不同位置发生的相关序列变化模式(绿色和黄色星号突出显示成对的比对列酸电荷和大小交换)(步骤1)。
- 目标序列和多序列比对形成了预测局部骨架结构的基础,包括扭角(显示了phi和psi预测,红色误差条表示不确定性)和二级结构(显示了步骤2;显示了PSIPRED预测)。还可以组装从预测具有相似局部结构的蛋白质中提取的骨架片段文库,以用于模型构建。
- 基于比对列中相关突变的观察,多序列比对可用于预测可能在空间上接触的残基对(步骤3)。
- 这些对局部结构和残基接触的预测使用诸如基于梯度的优化,距离几何或片段组装之类的技术指导了3D模型的建立(步骤4;显示了Rosetta42片段组装轨迹的快照)。
- 最初的3D模型通常以简化的表示和粗粒度的能量函数构建;为了更好地确near-native的预测,这些模型使用全原子能量函数进行了精炼,并相互比较以识别相似的低能构象簇,从中选择代表性模型作为最终预测(步骤5; 2D)显示了精炼模型空间的主成分投影,其中每个点代表一个模型。
2.2.1 片段组装 Fragment assembly
构象采样的一种流行方法是片段组装,其中,模型是从短而连续的骨架片段(通常长度为3-15个残基)中提取的,这些片段取自已知结构的蛋白质(图2)。选择此类片段的文库,通常针对目标蛋白的每个局部序列窗口为20–200,以提供可能的局部主链结构的采样。片段的选择通常由序列相似性以及局部结构特征(如二级结构或骨架扭转角)的预测指导。从这些片段构建全长3D模型采用蒙特卡洛模拟(box1),该模拟从随机或完全扩展的构象开始,然后通过重复选择蛋白质的随机窗口(例如22-30残基)并将其插入窗口从相应的片段库中随机选择的片段的结构。然后比较插入片段前后蛋白质模型的计算能量:如果插入片段后能量较低,则接受该移动,而如果能量较高,则接受片段插入的概率为随着能源的增加呈指数下降(都会标准,the Metropolis criterion)。为了生成假设模型,进行了数千次这样的模拟,每个模拟都包含数千个片段插入试验,最终形成了最低能耗的模型。片段装配模拟通常以简化的表示(例如,仅存在主链原子和单个“质心”侧链伪原子)和粗粒度的能量函数进行,该函数可以快速评估并定义相对平稳的能量适用于大规模构象采样的景观。然后,使用随后的原子详细细化模拟对候选模型进行排名并选择最终预测。
片段组装方法具有许多优点,有助于其普及:
- 首先,PDB中的几乎所有蛋白质结构在本地都与数据库中其他不相关的结构相似;
- 其次,使用经过实验验证的片段可确保模型通常具有蛋白质样的局部特征。
- 第三,片段库隐含地建模局部序列和结构之间的映射,而无需基础相互作用的准确能量模型。
- 第四,经验证明了片段装配模拟是探索蛋白质构象空间的有效方法,并且即使目标蛋白的某些局部区域在片段中的表达不充分,也能够对全局正确的折叠模型进行采样图书馆。在没有预测或实验确定的残基接触信息的情况下,片段组装方法通常在较小的α-螺旋或混合的α-β蛋白结构域上效果最佳,而全β和/或复杂的非局部拓扑结构则表现出最大的困难(由于片段插入动作会干扰远程接触)。
2.2.2 模型完善 Model refinement
区分原始状态的特征的高分辨率性质(例如,侧链堆积和氢键结合)意味着片段组装中使用的粗粒度分子表示(coarse-grained molecular representations )和能量函数不具有可靠选择near-native所需的精度,也不能用于填写基于结构的药物设计等应用程序所需的原子细节。因此,人们对模拟方法有相当大的兴趣,这些方法可以采用粗略的初始模型并将其移至更接近原始结构,同时通过消除诸如原子重叠或应变扭转角之类的特征来提高物理逼真度(physical realism)。此过程称为模型优化,需要精确的能量函数以及探索起始模型附近构象空间的策略。分子动力学模拟是一种已经成功应用于模型改进的构象采样策略(见box1)。在基于分子动力学的精炼中,将起始模型放置在由水分子围绕的模拟框中,然后根据牛顿运动定律和势能模拟经过许多小步前进的分子系统的轨迹能量函数。在最近的一种方法中,使用全原子能函数确定了在分子动力学模拟过程中采样的改进模型,并将其作为后续分子动力学模拟的起点,以进一步增强构象采样。原子详细的蒙特卡洛模拟(见框1)结合了侧链旋转异构体采样和Rosetta软件包的能量最小化,也已成功用于模型优化。最近的CASP蛋白结构预测实验揭示了高分辨率模型改进的进展,几个小组证明了多个预测目标的模型质量有了实质性的改善。这些性能提高的部分原因是由于改进的参数化程序而导致了基础全原子能量函数的准确性提高。
2.2.3 残基协变的联系预测 Contact predictions from residue covariation
对具有已知结构的蛋白质的多序列比对的分析表明,与空间接触中的残基相对应的成对比对列通常倾向于显示相关突变的模式:当一个列中的氨基酸发生变化时,另一列中的氨基酸也发生变化可能会发生变化(请参见图2步骤1中的路线中标有星号的两列)。对齐列之间的这种协变量归因于需要保留有利的残基-残基相互作用(例如氢键或紧密堆积)的需求。推动无模板建模改进的一个关键趋势是,可以通过分析多序列比对中的相关突变来预测残基之间这些空间接触的准确性。预测准确性的提高受到两个因素的推动:由下一代测序和宏基因组学驱动的蛋白质序列数据库的增长(因为更大的数据库意味着更深的多序列比对,因此有更大的能力检测相关的突变),以及全球统计方法的发展,该方法可以将由于空间邻近性而导致的直接残留物耦合与间接耦合分开。早期尝试使用协方差来预测空间接触的方法是采用“局部”度量,例如相互信息,它们独立考虑每对对齐列;这些方法部分由于统计相关性的传递性而导致误报:如果位置A耦合到位置B,位置B耦合到位置C,则即使位置A和C也将显示出显着的耦合,即使他们没有physically interacting。在“全局”方法中,通过找到比对列之间最直接解释观察到的序列相关性的直接相互作用的集合,来构建整个多序列比对的概率图形模型。然后,假定的交互位置对应于模型中权重最高的直接交互。
从协变分析得出的预测残基-残基接触已显示出可大大提高针对具有许多序列同源物的靶标进行无模板建模的准确性。片段组装方法已得到扩展,可以在折叠函数使用的能量函数中包含基于协方差的距离约束。最近使用Rosetta软件包进行的一项研究使用了宏基因组计划项目中的蛋白质序列数据来生成600多个未知结构蛋白质家族的模型。 EVfold方法使用最初开发的软件工具来根据实验距离限制(例如,根据NMR或交联实验)确定结构,以从协变派生的接触建立3D模型。该方法已成功应用于水溶性和跨膜蛋白结构的预测,以及对内在无序蛋白的结构状态的研究。限制基于协变的预测方法的实用性的主要因素是它们对相对较深的多序列比对的依赖(例如,其中的“深”已定义为包含数量至少为长度的平方根的64倍的非冗余序列)。尽管宏基因组学计划增加了可用的同源序列数量,但它们对原核生物的偏见意味着许多真核生物特异性蛋白家族的序列覆盖范围仍然太小,无法进行准确的预测。在这些情况下,可以集成序列和结构信息的各种来源的新的机器学习方法提供了一种有前途的途径。
2.3 蛋白质结构预测中的机器学习 Machine learning in protein structure prediction
机器学习技术在蛋白质结构分析中的应用历史悠久。诸如神经网络和支持向量机之类的机器学习模型已被用于预测一维结构特征,例如主链扭转角,二级结构和残基的溶剂可及性。最近,机器学习应用程序的重点已经转移到2D功能上,例如参加-参加接触图和参加间距离矩阵。认识到接触图类似于2D图像(其分类和解释是深度学习方法的惊人成功之一),蛋白质建模人员已开始将深度学习应用于识别PDB中蛋白质序列和结构的模式。卷积神经网络(专栏2和补充图1)在图像分析任务中表现出出色的性能,使其成为预测蛋白质接触图的自然选择。如何最好地编码有关目标蛋白的信息以输入到神经网络的问题是一个活跃的研究主题。例如,彩色图像通常被编码为三个实数矩阵:所有图像像素的红色,绿色和蓝色通道的强度(方框2)。使用DeepContact和RaptorX-Contact之类的方法作为输入的方法具有N×N(其中N是目标蛋白序列中氨基酸的数量)残基-残基偶联矩阵的衍生方法,用于目标蛋白的协变分析(增强通过预测局部序列特征)。在DeepCov和TripletRes(Y Zhang,个人交流)方法中,目标蛋白质多序列比对中的更多信息以400种(标准氨基酸数的平方)形式提供给网络。 N×N个特征矩阵,每个矩阵对应于定义的一对氨基酸,给定矩阵中位置(i,j)的值是对频率或给定氨基酸对在对齐位置i和j的协方差(另请参见专栏2和补充图1)。然后,卷积神经网络的任务是集成大量特征以识别空间接触,它通过训练大量已知结构的蛋白质及其相关的接触图和多序列比对来完成。性能最佳的CASP13结构预测方法突显了在无模板建模中整合机器学习的重要性,这些方法都依赖于深度卷积神经网络,以各种方式预测残差接触或距离,预测主链扭转角度( backbone torsion angles)并对最终模型进行排名。

Box2 |蛋白质结构分析中的深层卷积神经网络。 人工神经网络包含一层或多层顺序连接的处理单元(神经元)层,这些层将输入特征转换为输出预测。网络的每个处理单元都将来自上一层连接单元的加权信号集成为一个非线性响应,并将该响应传递到下一层的下游单元。在训练网络时,要在处理单元之间的连接上安装权重,以最大程度地提高预测精度。深度神经网络具有许多内部处理单元层(数以万计),可以使它们执行输入特征的高度复杂的非线性转换。卷积神经网络的特征是非常具体的分层连接,其中每个单元通过权重矩阵从上一层的单元本地窗口(例如,对应于输入图像中的小像素正方形)接收输入。卷积滤波器(参见补充图1)。卷积神经网络中的各层由对应于从输入中提取的不同信号的多个通道组成。每个通道都是通过扫描整个前一层的单个卷积滤波器得出的。在图像分类中,网络中较早的通道(更靠近输入)可能会识别输入图像中的特定局部几何特征(例如边缘或斑点),而较晚的通道则对应于更高阶的图案(例如合成特征)。通常在网络训练过程中优化网络中的层数和信道数。 将神经网络应用于蛋白质结构时的主要挑战是识别适当的编码,这些编码会将有关目标蛋白质的信息转换为适合于输入网络的实值特征。对于残基间接触预测,其中所需的输出是接触预测的2D矩阵,一种方法是提供由多序列比对得出的残基-残基协方差评分的2D矩阵作为输入。通过从训练过程中馈入网络的已知联系图和协变量矩阵的许多示例中学习,网络能够识别蛋白质联系图的特征(例如,重复的二级结构联系),从而使其能够优化基于序列的输入预测。对此类网络中早期卷积过滤器的分析表明,不同的过滤器专门用于检测不同的交互模式(例如,β-发夹或β-α-β主题)。最近的工作表明,可以从多序列比对中为神经网络提供更多信息-400个氨基酸对的频率或协方差矩阵(400个有序氨基酸对各一个,见图),而不是单个协变基质—可以进一步改善氨基酸接触和距离预测
三、蛋白质设计
蛋白质设计经常被称为逆蛋白质折叠问题。不是寻找给定蛋白质序列的最低能量构象,而是目标是确定能稳定所需蛋白质构象或结合相互作用的氨基酸序列。尽管问题的性质相反,但蛋白质设计所需的建模工具与高分辨率结构预测所需的建模工具非常相似。具有能量功能以对不同侧链和主链堆积相互作用的相对有利性进行排序至关重要,并且必须具有可用于搜索低能量序列和蛋白质构象的采样协议。蛋白质设计工作可以大致分为两类:
- 在基于模板的设计中,天然进化蛋白的序列和结构被修饰以实现新功能。
- 在从头设计中,根据设计要求和理化约束条件,从头开始生成新的蛋白质骨架和序列。
采取每种方法都有令人信服的理由。从头设计为我们对蛋白质结构的理解提供了严格的检验,并允许创建序列不受设计目标无关的不受序列限制的异常稳定蛋白质。基于模板的设计非常适合创建具有新功能的蛋白质,因为该蛋白质的结构是预先定义的,并且通常可以重新利用模板中的官能团。在这里,我们将介绍这两种方法的最新进展,重点是用于生成新型蛋白质骨架,复合物和功能位点的分子建模策略。
3.1 从头蛋白质设计
在大多数从头蛋白质设计的项目中,研究人员首先选择要构建的蛋白质折叠或拓扑。在某些情况下,选择特定的折痕是因为有兴趣对这种折痕有更好的了解。在其他情况下,选择折叠是因为这将是在蛋白质中注入特定功能的良好起点。例如,β-桶形蛋白质可在桶内形成小分子的结合袋。一旦选择了所需的蛋白质折叠,就构建了采用目标折叠的多肽骨架模型。对于给定类型的折叠,蛋白质可以潜在地填充大量构象,但是这些构象中只有一小部分与形成折叠良好,热力学稳定的蛋白质相一致。从头蛋白质设计领域的许多最新工作都集中在开发构建可物理实现的蛋白质骨架的改进方法上,因为它们可以使氨基酸侧链之间紧密堆积,从而满足氨基酸的氢键潜力。蛋白质骨架(主要通过二级结构形成),骨架扭转角几乎没有应变。
与蛋白质结构预测一样,一种用于确保一级序列中局部结构元件的适宜性的常用方法是,将天然存在的蛋白质的小片段组装成骨架(图3)。除了提供格式良好的二级结构外,这些片段还可以编码在二级结构的开始和末端在能量上有利的结构基序。为了创建采用所需三次折叠的主干,通常将基于片段的折叠算法与用户定义的距离约束相结合,以指定二级结构元素在3D空间中的定位方式。这种方法已被用于成功设计各种蛋白质折叠,包括所有螺旋蛋白质,重复蛋白质,混合的α–β蛋白质和仅由β-折叠和环组成的蛋白质。对于某些设计项目,蛋白质采用何种三级折叠可能并不关键,但是,蛋白质具有使其能够执行所需功能的特定结构特征可能很重要。例如,已经证明,将天然存在的螺旋-转-螺旋片段组装在一起的蛋白质可用于设计大量其他折叠,这些折叠具有诸如在蛋白质活性位点经常出现的口袋和凹槽等特征。诸如金属结合和蛋白质相互作用位点之类的功能基序也可以明确地并入装配过程中。

从小蛋白质片段构建理想折叠的过程通常会揭示有关物理和结构约束的新信息,这些约束决定了蛋白质可以采用的构象。采用果冻状折叠(由八个反平行β链形成的双链β螺旋)的全β折叠蛋白的第一个从头设计,显示出环之间的连接中环的几何形状和环的长度之间存在强耦合-张。类似的规则对于设计混合的α-β蛋白和α-螺旋蛋白中二级结构元素之间的连接也很有用。
用于生成从头蛋白质设计的骨架的另一种方法是建立一个数学模型,该模型使用少量参数来描述通常针对某种蛋白质折叠类型观察到的结构变异性。这种方法对于设计卷曲螺旋蛋白特别成功,该螺旋卷曲蛋白由围绕中心轴超螺旋的两个或多个α螺旋组成。使用最初由Francis Crick描述的盘绕线圈的数学(参数)模型,蛋白质设计人员可以快速生成大量的蛋白质骨架,其中每个模型都以系统的方式偏离下一个。例如,典型的是扫描超线圈半径(确定螺旋之间的距离)和超线圈扭曲(确定螺旋彼此紧密缠结)的替代值。这些研究表明,参数空间的某些区域允许螺旋之间更强的相互作用,可用于创建具有出色稳定性的蛋白质。参数设计的最新应用包括跨膜蛋白和α螺旋桶的从头设计。在此方法的扩展中,构建了一个参数模型来描述β-桶蛋白的骨架几何形状。有趣的是,这种方法没有成功,因为发现必须在骨架中包括非理想的扭结以减少扭转应变。最后,蛋白质片段的骨架组装更适合该问题,并允许从头设计β-桶。
3.2 基于模板的设计
对于许多设计目标,可能没有必要使用从头设计的蛋白质骨架和序列。相反,天然存在的蛋白质的高分辨率结构可以用作设计过程的起点。这种方法经常被用来设计与其他蛋白质结合的蛋白质,设计与配体结合的蛋白质以及设计酶。通常,将特定的蛋白质用作设计过程的起点并不重要,因为该项目的目标只是创建一种抑制蛋白质X或与配体Y结合的蛋白质。在这种情况下,考虑将大量天然蛋白质作为设计模板。每个模板将具有独特的结合口袋和分子表面,这将使某些模板比其他模板更适合设计目标。可以对模板进行预筛选,以找到具有适合目标配体大小的结合口袋的蛋白质,但是通常有必要使用多个模板进行设计模拟,以找到可以与目标分子形成紧密结合相互作用的结构。潜在的模板通常也经过预筛选,以选择稳定且易于生产的蛋白质。在某些项目中,从特定模板开始很有用或必要,因为它具有设计目标所需的功能。起始蛋白质可能是催化靶反应的酶,或是激活重要细胞表面受体的抗体。在这些情况下,设计目标通常是改善蛋白质的生物物理特性或以特定方式扰乱活性。
3.3 优化蛋白质序列 Optimizing the protein sequence
通过从头或基于模板的方法生成了蛋白质骨架的一个或一组模型后,设计过程的下一步是鉴定将稳定所需构象或结合事件的氨基酸序列。序列优化软件包括两个关键组件:
-
一个能量函数,用于评估特定序列的可取性,以及一个协议(protocol),用于搜索更有利的序列。由于在高分辨率结构预测和蛋白质设计(侧链堆积(side-chain packing),氢键,疏水性掩埋,主链和侧链应变)中需要优化相同的物理特性,因此经常将相似的能量函数用于设计和优化。通常使用各种基准来对能量函数进行参数化,这些基准致力于重现天然蛋白质的序列和结构特征。最近的一项令人振奋的进展是,证明可以通过同时优化能量功能以及小分子热力学数据和高分辨率大分子结构数据来改善蛋白质设计和蛋白质建模的能量函数。例如,训练能量函数以预测与将不同化学基团从水相移动到气相相关的实验测量的自由能变化,可以更准确地建模将蛋白质中的亲水基团和疏水基团掩埋的成本和收益。这种方法改善了传统蛋白质设计基准的结果,例如天然序列恢复(该方法旨在重新设计天然蛋白质时旨在产生类似天然的序列)以及通过突变预测自由能的变化以及改进在结构预测中。
-
设计模拟的第二个组成部分是寻找低能序列和侧链构象。已经针对该问题开发了多种方法,包括确定性方法(例如,末端消除和均值场优化)以及随机方法(例如,模拟退火和遗传算法)。大多数方法通过将侧链运动限制为在PDB的高分辨率结构中观察到的一组通常观察到的构象(或旋转异构体)来简化侧链优化问题。设计软件Rosetta将Monte Carlo采样与模拟退火结合使用,以识别低能序列和旋转异构体。从随机序列开始,Rosetta协议根据Metropolis标准接受或拒绝单个氨基酸突变或rotamer取代(这意味着可以接受降低系统能量的突变,并且可以提高系统能量的突变)。能量变化和模拟温度决定以某种概率接受系统;另请参见box1)。尽管这种方法很简单,但从不同随机序列开始的独立模拟会收敛于相似序列,并且该方法的优势之一是它的速度非常快(对于少于200个残基的蛋白质,通常在单个处理器上不到10分钟)。 诸如模拟退火之类的随机方法的局限性在于它们无法保证将确定最低能量序列。因此,研究团队还开发了可以识别给定蛋白质骨架和旋转异构体文库最低能量序列的方法。除了查找最低能量序列外,蛋白质设计软件包OSPREY还提供了对最低能量序列进行排名的方法
计算蛋白质设计的一个挑战是蛋白质的最佳氨基酸序列可能对蛋白质的精确3D结构非常敏感。在许多情况下,主链构象的微小变化会导致蛋白质能量急剧下降,而对最有利序列的较大变化。能量的这些大变化反映了化学相互作用的强度-原子坐标中很小的位移(<1Å)会导致强烈的空间排斥或诸如氢键之类的良好相互作用的丧失(图1b)。为了解决这种rugged energy landscape,蛋白质设计人员开发了多种方法来执行骨架采样以及序列优化。在设计从头蛋白质时,一种行之有效的方法是在基于旋转异构体的序列优化和基于梯度的最小扭转角(主干和侧链)之间进行迭代(另请参见box1)。通过这种方法,可以同时优化整个结构的角度。另一种方法是将蛋白质主链构象的离散变化与侧链旋转异构体取代偶联在一起。这些耦合的动作允许主链在计分变化的有利性之前调整为氨基酸取代
因为基于蛋白质全原子模型的能量计算对骨构象的微小变化非常敏感,并且因为用于对蛋白质构象评分的能量函数是经验模型,无法完全捕获所有有助于蛋白质能量的现象。对于蛋白质,不依赖于氨基酸侧链的显式建模的基于知识的序列设计方法也得到了发展。在一种方法中,将蛋白质分为空间上相邻的残基集,然后在PDB中搜索处于相似结构环境(称为三级结构基序,tertiary structural motifs,即TERMS)的残基。然后,使用PDB中TERM中每个残基位置的序列首选项来推导用于序列优化的能量函数。尽管使用天然蛋白质来获得序列偏好,但这种方法也可能适用于从头蛋白质设计,因为有限数量的TERMS(数百个)能够描述天然蛋白质中观察到的大多数局部结构环境
许多设计方案都受益于专门的序列优化方案。例如,使用逐步旋转异构体优化设计掩埋氢键网络(这对于蛋白质-配体和蛋白质-蛋白质的相互作用特别重要)是一项艰巨的任务,因为在所有极性基团都具有一个键之前,该网络在能量上是不利的氢键伙伴。换句话说,从没有氢键网络的序列中获得具有完全连接的网络的序列,要求序列优化模拟经过具有高能量的中间序列。为解决此问题,开发了一种使用潜在氢键伙伴(hydrogen bond partners)图形表示的侧链采样协议,以枚举输入主链结构的所有可能的侧链氢键网络。该方法允许设计具有大量埋藏的极性氨基酸和蛋白质链之间的高结合特异性的螺旋低聚物。创建这样的网络对于设计新酶至关重要,因为活性位点残基经常受到广泛的氢键的支持。另一个常见的设计问题是希望一种结合相互作用(称为阳性设计)胜于另一种结合相互作用(称为阳性设计)作为否定设计)。为了同时进行正向和负向设计,已经开发了多状态设计算法,该算法可以评估氨基酸构象的替代构象,并明确搜索会增加两个状态之间能隙(energy gap)的序列。
3.3 验证计算预测 Validating computational predictions
蛋白质设计项目中最激动人心的时刻是进行实验验证时。通常使用各种实验来表征设计的蛋白质,包括变性实验的稳定性分析,低聚状态的测量,高分辨率结构的确定以及功能分析(如适用)。对于许多设计目标,有必要通过实验表征一组替代设计,以识别少量成功的设计。根据设计目标,需要特征化的设计数量可以有很大的不同。稳定蛋白质的方法(另请参见下文)已发展到通常仅需表征少数设计的程度,而更具挑战性的问题(例如重新设计蛋白质的表面以结合另一种蛋白质)可能需要筛选数千种替代序列。在蛋白质设计项目中,通常要经历多代设计,然后每一代修改设计和/或计算策略。这样,就有可能了解实现设计目标所需的最小结构特征。在此过程的一个极好的例子中,基于酵母表面展示的高通量筛选策略与下一代测序相结合,以同时评估数千种从头设计的蛋白质。反复调整设计方案和筛选的过程导致产生了更稳定产生折叠蛋白的管道。
四、蛋白质设计的应用
计算蛋白设计的巨大前景之一是,它将允许创建在医学,工业和研究中具有有价值应用的新蛋白。 在过去的十年中,这一承诺已开始实现。 通过改进的计算方法以及DNA合成和测序技术的进步,可以实现这一进展。 现在,订购大批计算设计的序列可负担得起,从而使蛋白质设计人员可以快速探索问题的多种解决方案。 为了更好地说明可以应用计算蛋白质设计的问题类型,我们重点介绍了过去几年中蛋白质工程项目的子集,这些项目已将分子建模作为设计过程中的重要步骤。
4.1 稳定蛋白质 Stabilizing proteins
长期以来,人们一直在使用基于计算机的方法来鉴定突变,这些突变将增加蛋白质的热力学稳定性,因为这通常会导致重组蛋白的表达水平更高,并可能降低聚集的倾向。多年以来很明显,天然存在的蛋白质通常没有针对稳定性进行优化,并且对蛋白质进行整体重新设计(一种模拟,其中蛋白质中的所有残基都可以突变)可以显着提高热解折叠温度,在某些情况下超过30°C。但是,在大多数实际应用中,最好通过选择更具选择性的一组突变来提高稳定性。一种被证明非常适合此任务的方法是,通过在NCBI非冗余蛋白数据库中鉴定目标蛋白的同源物而生成的多序列比对中,将来自计算设计模拟的信息与序列偏好结合起来。许多研究小组证明,在该位置上用最优选的氨基酸替换在多序列比对中不能很好表达的氨基酸通常可以提高蛋白质的稳定性。为了减少这种类型的预测中的假阳性,并确定最有可能显着提高稳定性的突变,可以将来自多序列比对的信息与Rosetta设计模拟一起使用。在此方法的惊人展示中,一个步骤的设计过程足以识别出18个突变,这些突变将疟疾入侵蛋白RH5的耐热性提高了15°C以上,同时保留了所需的配体结合和免疫原性疫苗开发(图4a)。
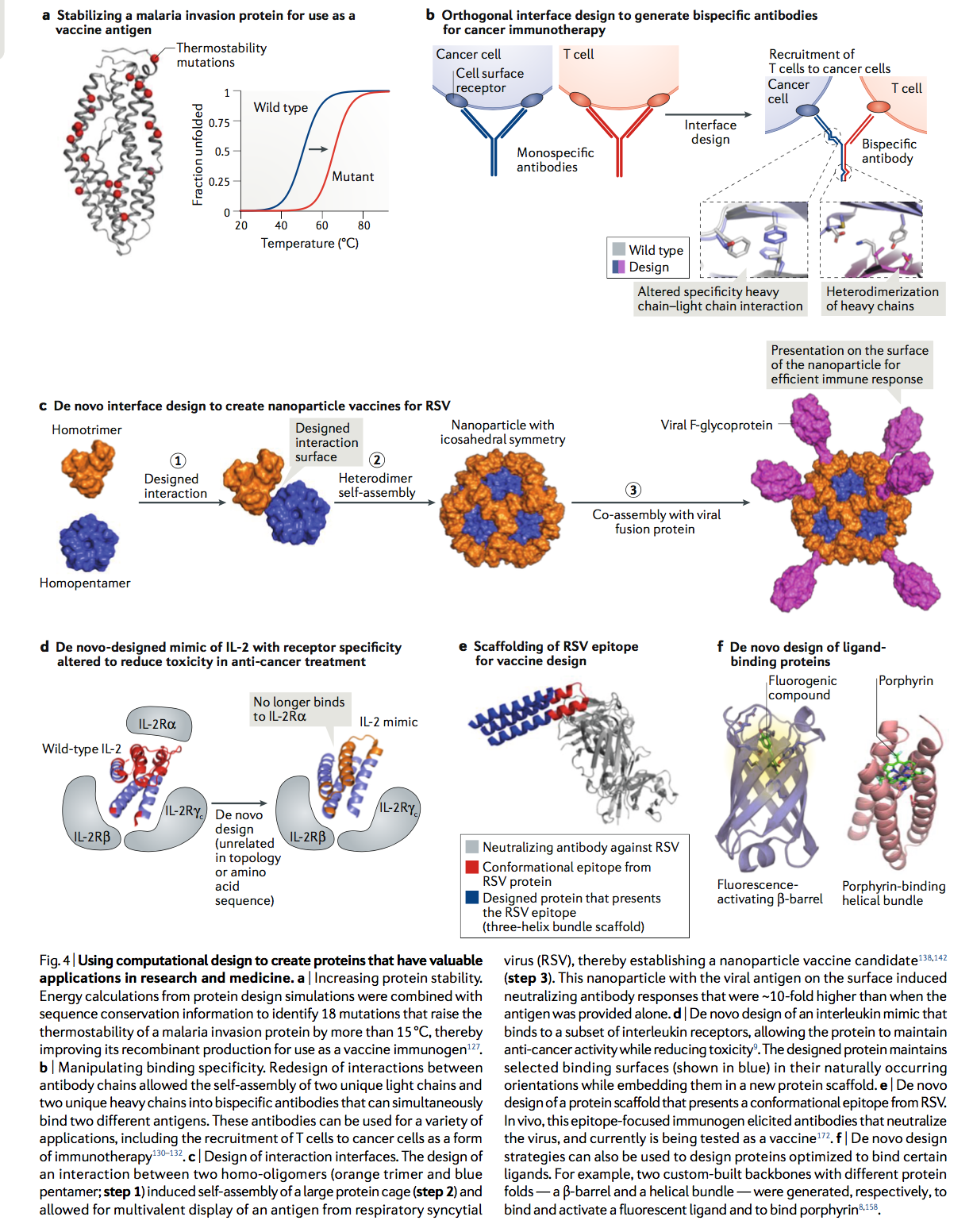
图4 |使用计算设计来创建在研究和医学中具有有价值应用的蛋白质。
a. 增加蛋白质稳定性。将蛋白质设计模拟中的能量计算与序列保守性信息相结合,以鉴定出18个突变,这些突变将疟疾入侵蛋白的热稳定性提高了15°C以上,从而提高了其重组生产量,可用作疫苗免疫原。 b. 操纵结合特异性。通过重新设计抗体链之间的相互作用,可以将两条独特的轻链和两条独特的重链自组装成可以同时结合两种不同抗原的双特异性抗体。这些抗体可用于多种应用,包括以免疫疗法的形式将T细胞募集至癌细胞。 c. 交互界面的设计。两种同型寡聚体(橙色三聚体和蓝色五聚体;步骤1)之间相互作用的设计诱导了大蛋白笼的自组装(步骤2),并允许呼吸道合胞病毒(RSV)的抗原多价展示,从而建立纳米疫苗候选者(步骤3)。这种在表面带有病毒抗原的纳米颗粒诱导的中和抗体反应比单独提供抗原时高约10倍。 d. 从头设计白细胞介素模拟物,该白细胞介素模拟物与白细胞介素受体的一个子集结合,使该蛋白质在降低毒性的同时保持抗癌活性。设计的蛋白质将选定的结合表面(以蓝色显示)保持其自然发生的方向,同时将其嵌入新的蛋白质支架中。 e. 从头设计蛋白质支架,该支架呈现RSV的构象表位。在体内,这种针对抗原决定簇的免疫原引发了中和病毒的抗体,目前正在作为疫苗进行测试。 f. 从头设计策略也可以用于设计优化结合某些配体的蛋白质。例如,分别生成了两个具有不同蛋白质折叠的定制骨架(β桶和螺旋束),以结合和激活荧光配体并结合卟啉(porphyrin)。
4.2 控制结合特异性 Controlling binding specificities
调节蛋白质相互作用的特异性是研究和重定向细胞信号通路的有效方法。改变的相互作用界面也可以用于组装新颖的分子组装体。在这种能力的一个证明中,多状态设计仿真被用来重新设计抗体重链和轻链之间的接触,以创建不再与野生型对应物相互作用的抗体变异体。这些重新设计允许创建双特异性抗体,其中IgG样抗体的一个臂识别一个抗原,而另一臂识别单独的抗原。这种类型的抗体在治疗上很有用-例如,当双特异性抗体可以将患者的T细胞募集到其癌细胞中时,作为一种抗癌免疫疗法(图4b)。操纵相互作用特异性的另一种方法是扩展相互作用界面,以设计目标蛋白区域中其他潜在结合配偶体之间不保守的新接触位点。该方法用于为存活前BCL-2蛋白家族的六个成员设计新颖的特异性抑制剂。这使研究人员能够探究每个家族成员在人类癌细胞系中生存信号转导中的作用。
4.3 从头界面设计 De novo interface design
为了设计两种蛋白质之间的新型相互作用,必须首先创建一个模型,使蛋白质彼此靠近,以使它们的表面相邻。使两种蛋白质彼此靠近的计算过程通常称为对接。一旦蛋白质相邻,可以使用序列优化模拟来搜索将稳定相互作用的氨基酸。由于可能无法设计许多对接的构象-例如,由于残基的位置不适当,无法实现高密度的有利相互作用,因此,考虑大批交替对接的复合物非常重要。特别成功的一种从头设计界面是创建对称的均聚物(symmetrical homo-oligomers)。这种设计方法有几个固有的优点:
- 通过对称复制有利的相互作用;
- 对接过程中采样的自由度降低;
- 由于不需要将蛋白质以未结合形式溶解,因此主要通过界面处的疏水相互作用可以稳定专一的同型低聚物。
通常,计算设计方法在设计非极性相互作用方面比在极性相互作用方面更成功。对称低聚物设计的一个引人注目的例子是自组装纳米笼的产生(图4c)。这些设计包括同二聚体或同三聚体的复合物,在复合物之间具有设计的界面以稳定纳米笼的形成。重要的是,只需要设计一个新的接口即可将其自动组装到机架中。这项技术的激动人心的应用包括包装RNA的de novo笼子,类似于病毒衣壳,通过冷冻EM有序呈递小蛋白进行结构测定,以及在纳米颗粒疫苗中展示病毒抗原(图4c)。还使用不需要创建新的蛋白质-蛋白质界面的方法设计了蛋白质笼。与如何使用DNA碱基配对组装大分子复合物(DNA折纸,DNA origami)相似,形成卷曲螺旋二聚体的螺旋可用于驱动蛋白质笼,而天然存在的同型寡聚体之间可使用工程刚性接头。有利于形成对称的保持架。对称相互作用的设计也已用于工程化有序的晶体状晶格和蛋白质,它们聚合成2D薄片和细丝。
单面界面(One-sided interface)设计(其中只有一种蛋白质伴侣发生了突变)可用于工程化结合和调节与疾病有关的信号蛋白的蛋白质。 但是,迄今为止,事实证明,基于计算机的单面界面设计比双面设计更加困难。 单面设计通常需要设计更多的极性接触,因为被靶向的蛋白质表面无法突变,并且可能包含一些极性氨基酸。 成功的单面设计通常与目标蛋白表面暴露的疏水斑块结合,就像结合和中和流感病毒的蛋白设计一样。 由于单面界面设计非常具有挑战性,研究人员已利用DNA合成和酵母表面展示的最新进展来实验筛选大量(数万个)计算设计的序列,以找到紧密,稳定的结合物。
一种特别难以捉摸的单面界面设计是基于计算机的抗体设计,该抗体与结合伴侣上确定的表面紧密结合。 这是一个具有挑战性的问题,因为除了设计跨抗体-抗原界面的相互作用外,还必须将抗体可变环稳定在有利于结合的构象中。 这些环可以访问广泛的构象,但是在抗体中自然发生的亲和力成熟过程中,它们通常会进化为采用独特的构象。 使用PDB的抗体环构象文库,创建具有确定的环结构的稳定抗体的最新进展令人鼓舞, 但尚无关于通过高分辨率结构验证的设计抗体与结合伴侣之间从头界面的报道。
4.4 支架蛋白结合位点 Scaffolding protein binding sites
对于一些涉及蛋白质-配体或蛋白质-蛋白质相互作用的项目,已经知道有利于结合的结构和序列基序,但是将结合基序支架在折叠良好的蛋白质中将很有用。通过对基序进行构架,可以将其预先排列为具有结合能力的构象,从而增加结合亲和力,并可以提高蛋白质的稳定性和溶解性。上述的BCL-2蛋白抑制剂的设计就是一个例子。在另一个例子中,创建了天然细胞因子IL-2和IL-15的模拟物,它们表现出仅与白介素受体亚组相互作用的结合表面(图4d)。从IL-2中选择的对于结合所需的白介素受体子集非常重要的螺旋以一种使螺旋适当定位以结合其受体的方式并入从头设计的蛋白质中。像天然存在的IL-2一样,设计的蛋白质在用于黑色素瘤和结肠癌的小鼠模型中时具有抗癌活性,但对小鼠的毒性较小。母体嫁接也已用于展示构象表位,用于疫苗开发。这种方法已显示出有望引发幼稚B细胞对源自HIV和呼吸道合胞病毒(RSV)的支架抗原的反应(图4e)。值得注意的是,除了增加疫苗生成所需的免疫原性外,蛋白质设计还可用于使蛋白质的免疫原性降低。例如,设计模拟已用于搜索蛋白质突变,该突变将保持稳定性和活性,但去除已知为主要组织相容性复合物(MHC)的良好底物的序列基序。
4.5 设计的配体结合和催化 Designed ligand binding and catalysis
在现有或从头设计的蛋白质中引入新的配体和底物结合位点引起了极大的兴趣,因为这可用于创建新的成像试剂,催化剂和传感器(图4f)。在这种类型的设计中,围绕蛋白质口袋的氨基酸侧链是可变的,以便与所需的配体形成有利的接触。在许多情况下,特定的蛋白质结构可能没有适当定位以与所需配体形成强相互作用的残基。为了实现更紧密的接触,可以通过对蛋白质主链进行小的改动来调整结合口袋,或者可以将其他蛋白质视为设计的起点。从头设计方法为创建一组蛋白质结构提供了一种策略,该结构可以通过计算筛选出有利的结合口袋。在最近的一项研究中,一组从头设计的β-桶被用来结合荧光分子,其构象可增强荧光发射,可用于细胞成像。结合位点设计还可用于预测突变,从而导致对治疗剂的耐药性。
催化剂的从头设计特别具有挑战性,因为酶不仅必须结合底物,而且还必须稳定过渡态并释放最终产物。已经开发出一种计算方法,用于快速扫描一组蛋白质结构,以找到适当定位以构建酶活性位点的残基群。一旦关键的催化残基就位,就在催化残基周围的残基(第二壳层)上进行进一步的侧链设计,以使催化残基稳定在所需的构象中。这种方法取得了一定程度的成功,但是到目前为止,合理设计的酶还没有达到与天然酶相当的催化能力。由于目前的方法无法以足够的精度放置和/或稳定活性位点残基(可能需要以亚埃级精度进行设计,而许多计算设计仅在1或2 Å)或因为高效酶具有许多其他功能,例如底物和产物快速进入和退出结合位点的机制。
4.6 蛋白质开关的设计
蛋白质的一个重要特性是它们经常在其他构象之间切换。这种转换是多种蛋白质功能不可或缺的,包括信号转导,分子马达和催化作用。采用替代主链构象时,多状态设计仿真可用于搜索低能序列。这种方法已经被用来设计一种可以在不同折叠之间切换的小蛋白,并且可以设计一种跨膜蛋白,这种蛋白在不同构象之间摇摆以将金属跨膜运输。在此策略的扩展中,多状态设计用于将功能相关时标(毫秒)发生的构象转换引入现有细菌蛋白中。分子建模还可以用于辅助设计将天然存在的构象开关与功能性基序结合在一起的嵌合蛋白。此策略已用于创建可光活化的蛋白,以控制活细胞中的寡聚和催化作用。蛋白质设计软件还可用于告知蛋白质中可光活化化学基团的位置,以控制构象转换。
五、结论与观点
在过去十年中,用于蛋白质结构预测和设计的工具取得了长足的进步,但仍然存在许多挑战。指导预测和设计的能量函数(出于计算效率的原因,必须近似)仍难以准确平衡极性和非极性相互作用以及溶剂化效应,尤其是在界面处。结果,接口建模(interface-modelling)应用的成功率仍然很低,例如具有主链灵活性的蛋白质对接,单面接口设计和酶设计。明确地模拟“结构上重要的”有序水分子子集并与建模的蛋白质表面进行关键相互作用的混合方法代表了一种有前途的途径,但是由于其计算成本以及平衡相互作用与显性分析的需要,这些方法仍然难以进行参数化水和大量水被隐式建模。环介导的相互作用,例如T细胞受体与MHC肽或抗体和抗原之间的相互作用,也仍然难以预测和工程化。在这里,界面能量的挑战因需要精确建模不规则多肽片段的构象偏好而变得更加复杂,这些片段可能会采样未结合状态的结构整体。更广泛地讲,需要新的方法来稳健地预测和设计通常对蛋白质功能至关重要的蛋白质构象柔韧性和运动。可能需要将分子动力学轨迹与能量分布图分析相结合的方法来捕获这些灵活系统的动态方面。
尽管存在这些挑战,但我们认为蛋白质预测和设计方法随着它们的不断成熟,在生物学和医学中将发挥越来越重要的作用。天然存在的蛋白质的一个显着特征是,为了发挥其在细胞中的作用,它们经常参与多种活动和结合反应。例如,Rho家族的GTPases催化GTP的水解,经历明确的构象变化,与多种下游信号蛋白结合,并受到愈创木瓜交换因子和鸟嘌呤激活蛋白的调节。我们预计,通过将定向进化方法与分子建模的进步相结合,具有相似复杂性的蛋白质的设计将很快实现,并将实现各种激动人心的应用。例如,从头设计的蛋白笼(protein cages)可能会在疫苗开发中用作计算机上设计的免疫原的支架,并在癌症治疗中作为“智能”药物传递载体,能够整合多个靶向提示(例如,异常细胞表面蛋白表达或MHC呈递的新表位)。蛋白质的工程控制机制(配体结合,光激活,酶激活等)应允许设计仅在特定环境中具有活性并减少毒副作用的治疗剂。
随着蛋白质结构数据库的不断增长,新的蛋白质骨架和侧链堆积安排的可用性将增加,从而为重新利用它们创建新的结合位点和功能提供了可能性。就像机器学习和结构预测一样,看看机器学习和模式识别(pattern recognition)是否可以促进蛋白质设计领域也将令人兴奋。
参考资料
- Computational Biology Program, Fred Hutchinson Cancer Research Center, Seattle, WA, USA. 4Institute for Protein Design, University of Washington, Seattle, WA, USA. https://doi.org/10.1038/s41580-019-0163-x. Advances in protein structure prediction and design
