【4.5.4】使用Robetta服务器进行蛋白质结构预测和分析
Robetta服务器( http://robetta.bakerlab.org )提供了用于蛋白质结构预测和分析的自动化工具。对于结构预测,将提交给服务器的序列解析为推定域,并使用比较建模或从头结构预测方法生成结构模型。如果使用BLAST,PSI-BLAST,FFAS03或3D-Jury找到了与已知结构的蛋白质的可靠匹配,则将其用作比较建模的模板。如果找不到匹配项,则使用从头开始的Rosetta片段插入方法进行结构预测。实验性核磁共振(NMR)约束数据也可以与RosettaNMR从头结构确定的查询序列一起提交。当前的其他功能包括使用计算界面丙氨酸扫描预测突变对蛋白质相互作用的影响。 Rosetta蛋白质设计和蛋白质-蛋白质对接方法也将很快通过服务器提供。
一、介绍
Robetta是一项互联网服务,提供自动结构预测和分析工具,可用于从基因组数据推断蛋白质结构信息。服务器使用第一个全自动结构预测程序,该程序会在与已知结构的蛋白质存在序列同源性的情况下,为整个蛋白质序列生成模型。
- Robetta使用比较建模将输入序列解析为域,并使用与已知结构的蛋白质具有序列同源性的域建立域模型,并使用Rosetta de novo结构预测方法为缺乏此类同源性的域建立模型。结果给出了跨越全长查询的模型的域预测和分子坐标。
- 服务器还可以利用用户提供的核磁共振(NMR)约束数据,使用RosettaNMR(1-3)协议确定蛋白质结构。这些工具可以与当前的结构基因组计划一起使用,以帮助加速结构确定并获得针对目标开放阅读框(ORF)的结构见解。
- 另外,由于多结构域蛋白通常难以结晶且许多蛋白对于NMR结构确定而言太大,因此使用Robetta进行结构域预测可通过扩大可从其确定结构的靶标库来帮助结构基因组学。
- 致病性原生动物的结构基因组学( SGPP; http://www.sgpp.org )联盟目前正在使用内部版本的Robetta来识别表达并从不表达为完整链的ORF结晶的片段,并辅助结构的细化。
- Robetta还提供了使用“计算界面丙氨酸扫描”来鉴定蛋白质-蛋白质复合物界面中涉及的能量重要侧链的功能(4,5)。
Robetta的最终目标是提供足够质量的结构信息,以协助研究,推断功能和协助药物设计。比较模型已经用于推断功能并指导实验工作,整个研究领域正在不断完善,如“结构预测的关键评估”(CASP-5和“全自动”的CAFASP-3)实验所示( 6,7)。罗贝塔(Robetta)在这些评估中表现最好。
二、方法
Robetta使用Rosetta软件包的全自动实现来预测蛋白质结构。 参考文献(7-9)中详细描述了Rosetta方法,参考文献(6,7)中描述了Rosetta在CASP-5和CAFASP-3中的使用。
2.1 Domain prediction
为了预测全长蛋白质序列的结构,Robetta使用称为“ Ginzu”的域预测方法(6)作为结构预测的初始步骤。 Ginzu是一种分级筛选程序
- 首先使用BLAST,PSI-BLAST(10),FFAS03(11,12)和3D-Jury(13,14)来检测查询序列中与实验确定的结构同源的区域,然后继续使用基于多序列比对(MSA,multiple sequence alignment )的方法来预测推定的结构域(图1)。按照每种方法的可靠性对过程进行排序,从最可靠的方法(BLAST)开始,然后是根据置信度级别(PSI-BLAST)的下一种方法,依此类推。如果找到匹配项,则序列中剩余的不匹配部分将用作下一步的输入。
- 使用我们的比较建模协议对与具有已知结构的序列同源的区域进行建模。如果未分配的区域少于50个残基,则将其视为域连接子(linkers),或者使用HMMER(16)在Pfam-A(15)中搜索可能为域的区域。
- 最后一步尝试通过使用针对NCBI非冗余(NR)蛋白序列数据库的PSI-BLAST搜索获得的全长靶标的MSA,来识别其余未发现序列中的推定域。将MSA中频率最高的非重叠序列簇分配为域,并在序列末端发生率高,使用PSIPRED进行强loop预测(17)并减少序列对齐残基的占有率。弱的位置偏好也用于在未分配区域的中间附近放置切口。来自Pfam-A的推定域和基于MSA的方法被标记为从头结构预测。甚至可以进一步解析这些域,以满足我们从头协议的大小限制(约200个残基限制)。 PDB(蛋白质数据库)中的大部分域都在此长度之内(18)。
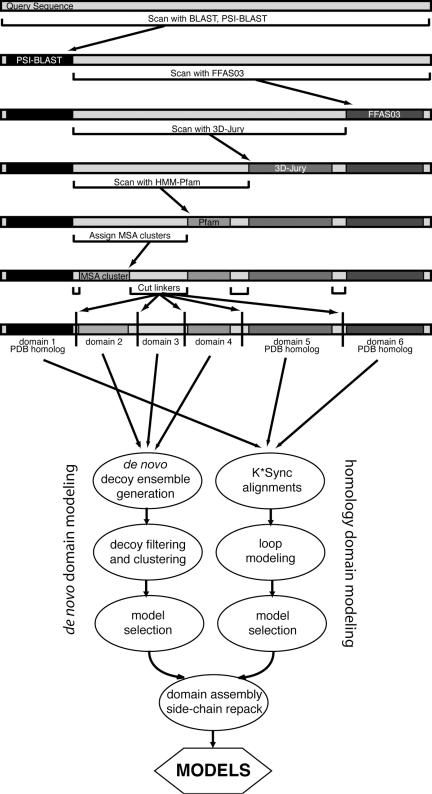
图1 Ginzu域解析层次结构和建模协议(protocol)。 扫描查询序列,以查找与可能是域的已知结构和区域的匹配。 未覆盖但足够大以成为一个域(至少50个残基)的部分将作为下一步的输入传递。 该顺序基于每个步骤的相对精度。 首先执行同源结构搜索,然后针对Pfam-A进行搜索,然后根据MSA序列簇进行解析。 分配了边界,以使没有同源结构的推定域在Rosetta de novo协议可访问的大小限制之内。 与具有已知结构的序列同源的区域用于比较建模。 对Pfam-A,MSA群集域和足够大小的其余未覆盖区域的匹配进行从头结构预测。 在最后一步中,将领域模型组装为完整模型。
2.2 比较建模 Comparative modeling
比较建模方法在以前发表的论文中有更详细的描述(6)。 Robetta并非简单地从用于检测结构同源物的方法中获得比对结果,而是尝试通过使用一种称为K * Sync(D。Chivian,准备中的手稿)的方法,利用残基图谱与图谱(residue profile–profile)比较来获得改进的比对结果。 二级结构预测以及有关必须折叠的元素的信息,以通过动态编程生成单个默认对齐方式(19)。 K * Sync还以参数方式生成一个对齐集合,从中选择诱饵模型。未对齐区域被视为循环,然后使用Rosetta片段插入方法(20)在固定模板的上下文中对其进行建模。使用包括缺口闭合项的能量函数以确保肽主链的连续性。首先组装短和中环(<17个氨基酸),将得分最低的一组环添加到模板中,然后构建长环。进行多次独立的模拟,并选择最低的得分构型作为适合给定比对的loop组合。使用Rosetta能量函数的不同变体从此组合中选择四个模型,并返回带有默认K * Sync对齐方式的模型。
2.3 从头开始结构预测与Mammoth
Robetta使用先前已经描述过的从头结构预测协议的稍微修改版本(6)。对原始方法进行了修改,以在合理的时间范围内运行公共服务器的查询。像原始协议一样,Robetta会生成三个和九个残基的片段库,这些片段库表示在PDB中看到的局部构象,然后使用支持蛋白质样特征的评分功能通过片段插入来组装模型。 Robetta为原始查询生成10 000个诱饵,为最多两个序列同源物生成5000个诱饵。然后,根据得分,并根据它们是否通过过滤器来消除具有过多本地联系或不太可能具有股线拓扑结构的诱饵(decoys)的筛选器,为每个同源物选择2000个查询诱饵和1000个诱饵。然后,基于所有未定位位置上的Cα均方根偏差(RMSD)对选定的诱饵进行聚类。排名最高的9个聚类中心被选为排名最高的模型,通过上述过滤器的最佳评分模型被选为第十个模型。然后使用Mammoth(21)对一组代表性的PDB链进行最终模型的搜索,以找到相似的结构,以鉴定与已知结构蛋白质的潜在相似性。Mammoth确定了模型与PDB中蛋白质之间最长的结构重叠,并报告了Z值(22),这表示偶然在大小相似的蛋白质之间获得相似长度匹配的可能性。这些搜索的结果将用于置信度函数中,该函数将在以后的版本中添加。
2.4 域组装和侧链包装 Domain assembly and side-chain packing
如果将查询解析为多个域,则结构预测过程的最后一步是将域模型组装为连续的全长结构。 Robetta使用以N端域开头的迭代域组装协议,并使用与我们从头协议相同的评分方法,尝试通过片段插入由Ginzu分配的推定linker区域进行域关联。如果链包含两个以上的域,则将第三个域添加到先前组装的模型中,然后过程继续进行直到组装整个链。尽管我们正在努力改进此方法,但是由于使用高分辨率晶体结构进行了基准测试,因此最后一步必须被视为一种美学处理,对于低分辨率模型而言,这可能是不准确的。一旦链被完全组装,最终模型的侧链将使用带有依赖于主链的侧链旋转异构体库(24)的蒙特卡洛算法(23)重新打包。
2.5 RosettaNMR
用户可以提供用于确定RosettaNMR结构的实验NMR约束数据。 已发表的论文(1-3)中介绍了RosettaNMR方法。 Robetta使用的协议与已发布的方法略有不同。 Robetta使用与RosettaNMR相同的方法来生成片段库,该库与化学位移,NOE约束数据以及(如果存在足够的数据)残余的偶极耦合相一致。 然后将片段库与相同的RosettaNMR de novo片段插入方法一起使用,该方法利用其评分功能中的约束数据生成诱饵。 但是,此时协议有所不同。 Robetta继续使用从头开始的聚类诱饵协议,并为最终模型选择聚类中心和评分最低的诱饵,而不是像RosettaNMR那样仅选择评分最高的模型,然后进行模型优化。 因此,Robetta的结果准确性可能会稍差。
2.6 界面丙氨酸扫描
Robetta包括“计算界面丙氨酸扫描”(4)方法,该方法可预测截短突变对蛋白质-蛋白质复合物稳定性的影响,如参考文献所述(4,5)。 简而言之,该程序可识别蛋白质-蛋白质界面中涉及的残基,并使用简单的自由能函数计算每个侧链被丙氨酸单取代后结合自由能的变化。 在19种蛋白质-蛋白质复合物中的233个突变的测试集中,正确预测了79%的残基具有重要的能量意义,而68%的中性残基得到了正确预测。
三、输入,输出和选项
3.1 注册
用户必须先注册( http://robetta.bakerlab.org/register.jsp ),然后才能将作业提交给Robetta。
3.2 结构预测服务器
提交给结构预测服务器的序列必须为一个字母的氨基酸格式。它们可以粘贴到提交表单中,也可以从文件中上传。用户可以选择提交序列以进行域识别或完整结构预测。用户还可以选择指定用于比较建模的PDB ID和链。
对于RosettaNMR提交,用户必须上传实验NMR约束数据(化学位移,NOE数据和/或残留的偶极耦合)。每种数据类型所需的输入格式在 http://robetta.bakerlab.org/documents/data_formats.jsp 中进行了描述。
通过单击队列表( http://robetta.bakerlab.org/queue.jsp )中列出的作业ID,可通过Web界面(图(图2)2)提供特定作业的结果。对于完整的结构预测,还会通过电子邮件将坐标发送给用户。为了进一步了解,将显示以下结果以及预测模型(图22A):
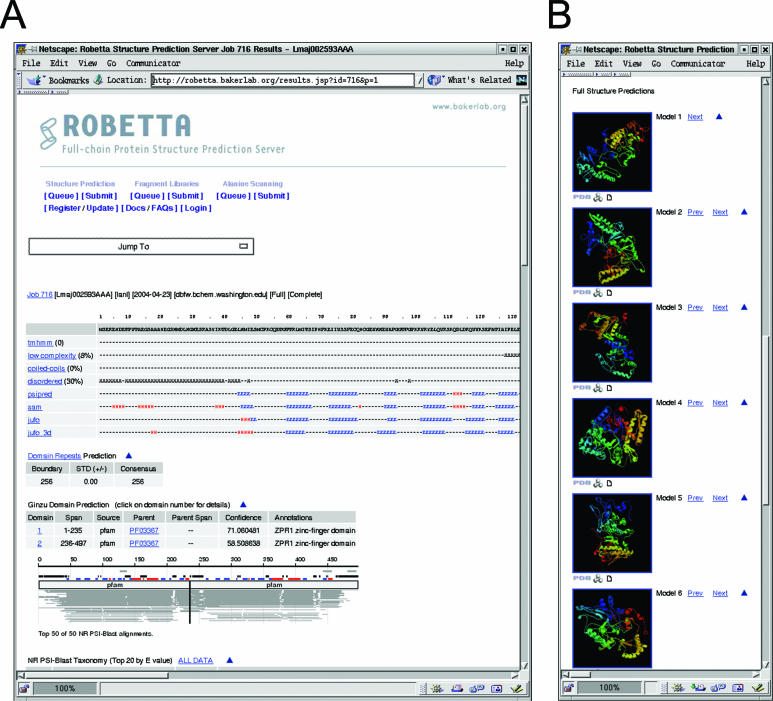
Figure 2 An example of structure prediction results. Screen shot of the top of the web page (A) and the first 6 of 10 structure predictions located at the bottom of the web page (B).
-
使用TMHMM预测跨膜螺旋(25,26);
-
程序SEG分配的低复杂度区域(27);
-
使用COILS进行的盘绕线圈预测(28);
-
使用DISOPRED预测无序区域(29);
-
使用PSIPRED(17),SAM-T99(30,31),Jufo和Jufo3D(32)进行二级结构预测;
-
上面列出的结果,域预测协议中用于域预测协议最后一步的域预测和NR PSI-BLAST多序列比对被浓缩为图像,以帮助确认域预测结果;
-
使用REPRO进行域重复预测(33,34); 如果检测到重复,则给出预测的边界;
-
最高的NR PSI-BLAST结果和由最低E值确定的前20个物种的注释。
完整查询的模型以图像显示在页面底部(图(图2B).2B)。 通过单击每个模型图像下方表示的图标,可以从网站上下载这些模型的坐标。
通过单击“ Ginzu域预测结果”表中列出的域名,还可以为每个域提供特定结果。 对于比较模型,将显示用于建模的K * Sync路线。 对于从头算模型,将显示Z分数> 4.5的前10个匹配项的Mammoth(21)结构模型比较结果。 可以通过单击Z分数下载实际的Mammoth结构模型比对,并使用RasMol等分子观察器进行观察以进行进一步检查。 用户可以通过单击每个域模型图像下方的图标来下载域模型。
3.3 界面丙氨酸扫描 Interface alanine scanning
关于如何使用Web界面进行计算界面丙氨酸扫描的详细说明和说明已经发布(4)。作为输入,用户必须提供蛋白质-蛋白质复合物的PDB坐标,并定义哪些链属于要扫描的结合界面的伴侣。用户还可以选择提供要考虑的界面残基列表。如果未提供列表,则使用接口中涉及的所有侧链。通过电子邮件将结果作为计算得出的每个所考虑的界面侧链的结合自由能变化(ΔΔGbind)的列表发送电子邮件。
3.4 片段库
Robetta包含一个片段库服务器,以容纳正在本地运行Rosetta软件包但无法生成片段库的研究小组。对于Rosetta片段库,用户必须以单字母氨基酸格式提交查询序列。 NMR约束数据也可以上传以生成片段库,以用于RosettaNMR。作业完成后,将通过电子邮件将下载结果的URL发送给用户。由于片段库很大(大约兆字节),因此作业完成后一周将它们从服务器中删除。
四、结构预测性能和局限性
Robetta的性能已在CASP-5和CAFASP-3实验中进行了评估,并使用LIVEBENCH( http://bioinfo.pl/LiveBench )不断进行基准测试(35)。在先前的出版物中已经讨论了Robetta在CASP-5和CAFASP-3中的性能(6,7)。总体而言,Robetta在同源性建模和折叠识别目标方面与其他服务器相比非常出色,并且在远程折叠识别和新折叠目标方面使用了从头协议,从而做到了合理的工作。 Robetta用于LIVEBENCH的比较建模协议的全面评估将在另一份手稿(D. Chivian,准备中的手稿)中讨论。表1.1总结了LIVEBENCH 7和8中Robetta在从头预测域上的性能。由于仍然无法高精度地进行从头开始的结构预测,因此我们在表中使用了两个相对宽泛的正确预测定义。首先,如果10个模型中的至少1个具有与原始结构的RMSD为4Å或更小的50个或更多残基的Mammoth比对,则认为域正确建模(定义I,表1的上半部分) ,1)或第二个Mammoth Z得分大于或等于天然结构(定义II,表Table1的下部).1)。在这两种定义下,Robetta可以为超过一半的结构域提供正确的预测,并且对于由全α或α-β二级结构组成的结构域尤其适用。还很好地建模了具有全β二级结构的结构,该结构包含反平行的β-折叠,且长度在151-200个残基范围内。由于域预测中的错误,残基<100的域通常不正确。
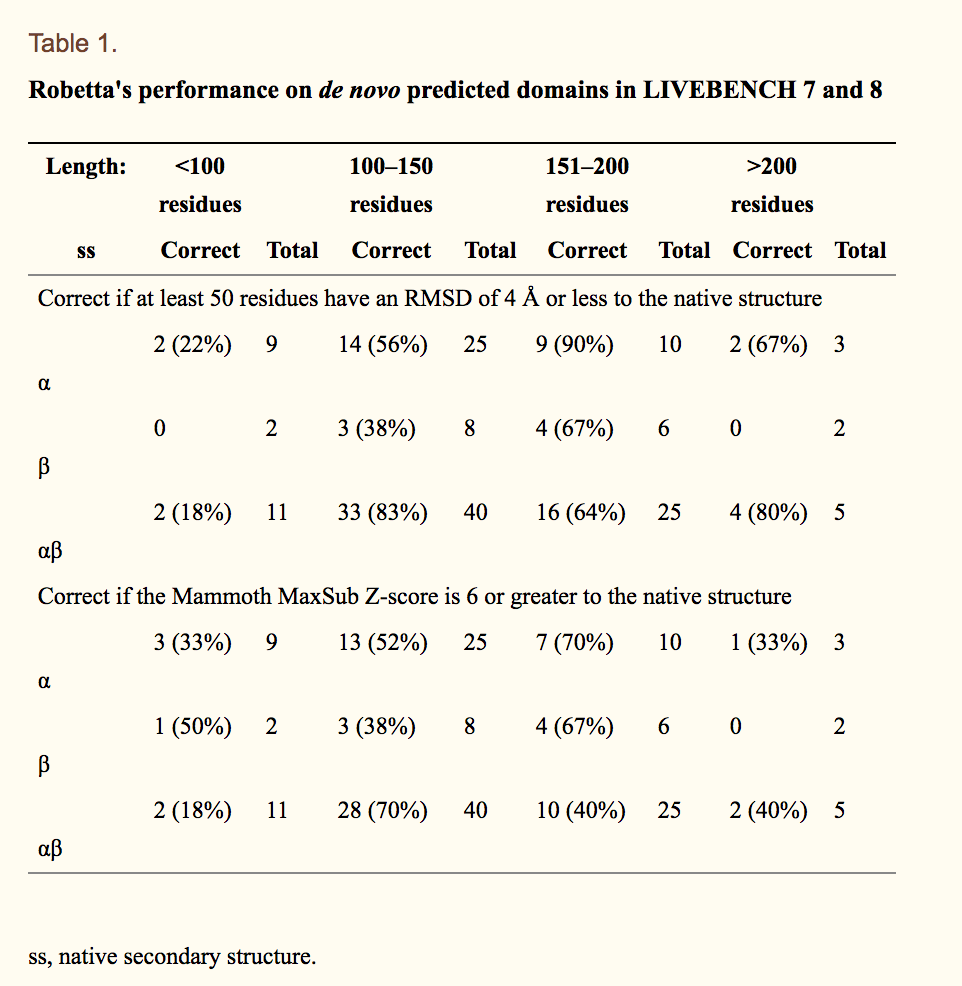
与所有结构预测方法一样,使用Robetta时要考虑许多限制。 从头协议已针对小型单域蛋白(<120个残基)进行了优化。 在此限制内,模型的RMSD通常约为3–7Å,超过原始结构的一半。 超过此限制,模型仍可能在4ÅRMSD内至少有50个残基,如表Table1.1所示。 对于比较建模,模型的质量很大程度上取决于对最佳父模板和对齐方式的正确选择。 由于这些因素,结果在很大程度上取决于域分配的准确性。 遵循的一般规则是,按照该顺序,应该将BLAST,PSI-BLAST,FFAS03和3D-Jury亲本检测视为最可靠。 从Pfam-A和MSA预测的域应谨慎对待,尤其是对于较长的域以及那些仅由MSA分配的域。
五、通透 THROUGHPUT
Robetta的计算要求很高,并且需要相当大的计算机集群才能在合理的时间尺度上进行结构预测。因此,为了满足需求,Robetta被设计为可以在计算机群集中运行,这些计算机群集可以作为镜像在区域内分布。 Robetta大约需要4到6小时才能在大约80个CPU的单个集群上运行150个残差查询。有了两个大小相似的群集的镜像,Robetta当前每天能够处理大约10个正常大小的作业。由于这种计算需求,用户一次只能提交一个序列到作业队列。提交作业以进行结构预测后,完成作业所需的时间取决于查询的时间长度,已排队的作业数和可用的CPU数。此时间以每项作业的天数估算,并列在队列表中,以使用户了解作业何时完成。在没有3D-Jury和片段库的情况下进行域预测的要求要低得多,并且在单个处理器上大约需要15-30分钟。计算丙氨酸扫描是一个快速的过程,只需几分钟即可在单个CPU上运行。
六、未来的工作
我们计划通过添加蛋白质设计(36)和蛋白质-蛋白质对接(37)功能来扩展Robetta服务器的范围,这些功能是在我们实验室中作为Rosetta软件包的一部分开发的。 我们还计划为从头结构预测结果添加置信度值,该结果源自与先前研究中使用的函数相似的函数(38)。 将添加比较建模选项,这些选项将允许用户提供自己的起始模板。 我们希望通过扩展群集镜像的分布式网络来提高Robetta的吞吐量。
参考资料
- Nucleic Acids Res. 2004 Jul 1; 32(Web Server issue): W526–W531. Protein structure prediction and analysis using the Robetta server. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC441606/
