【4.7.3.4】结构比较--Tm score (Lg score/maxsub score)
我们开发了一种新的评分功能,即模板建模评分(TM-score, template modeling score),以通过扩展全球距离测试(GDT, Global Distance Test)和MaxSub中使用的方法来评估蛋白质结构模板的质量和预测的全长模型。
- 首先,利用蛋白质大小依赖性量表来消除先前分数的固有蛋白质大小依赖性,并适当地考虑随机蛋白质结构对。
- 其次,不是设置特定的距离截止值,而是仅计算误差低于截止值的分数,而是在提议的分数中评估对齐/建模中的所有残基对。
为了比较各种评分功能,我们使用threading程序PROSPECTOR_3为1489个中小型蛋白质构建了大规模的结构模板基准集,并使用MODELLER和TASSER构建了全长模型。与GDT和MaxSub评分功能相比,初始threading对齐的TM得分显示出与最终全长模型的质量更强的相关性。 TM分数在最近的CASP5实验中被进一步用作对所有“新fold”目标的评估,并且与人类专家的视觉评估结果非常吻合。这些数据表明,TM-评分是对蛋白质结构预测的全自动评估的有用补充。可从 https://zhanglab.ccmb.med.umich.edu/TM-score/ 免费下载TM-score的可执行程序。
附件内容
很少有研究是完全新颖的,TM-score 也不例外,它其实是LG-score的改进MaxSub的改进。
1997年,Michael Levitt,这位日后的诺贝尔奖得主,与他的博后Mark Gerstein,目前耶鲁大学生物医学信息学、分子生物物理与生物化学、计算机学的三聘教授,出于统一比较蛋白序列相似度和结构相似度的想法,提出了Levitt-Gerstein score,也就是 LG-score(公式12)。[6] LG-score 是首个不依赖于序列的结构对齐打分。
2000年,Siew等人提出 MaxSub 算法,包括一种结构重叠对齐算法和一种基于 LG-score 的结构相似度打分(公式13)。[7]
2004年,张阳与导师 Skolnick设计出template modeling score,也就是 TM-score(公式14),评估全长模型的预测,消除蛋白质的大小对结构打分的影响;更重要地,TM-score 可判断2个相比较的结构是否属于同一 fold,即在整体结构或拓扑层级评价结构。[5]

相对于 LG-score,MaxSub 去掉了未匹配残基的空位罚分(gap penalty),并且去掉了无用的常数 M,代之以 1/LN,从而在优化过程中需要最大化地重合两个比较结构的子结构,只考虑对齐后 di ≤ d0 的残基,尽可能地增加 LT;相对于 MaxSub,TM-score 则将常数 d0 替换为一个 LN 的函数,从而使得整体打分没有链长依赖。
理解 TM-score 的物理含义以及它相对 LG-score 和 MaxSub 的改进,关键在于理解公式14中的 d0 —— d0 是天然结构的长度(LN)的函数,可是为什么令 di 除以 d0 就能消除链长依赖?
注意公式14中 di 的定义,它其实就是单个残基的 RMSD,它的均值我们在公式8中已经给出。因此,di 的均值可以由公式15近似表示。作者发现,令常数 h = 0.75,则 di 的均值可以进一步用更简单的函数形式近似,即公式14中的 d0。
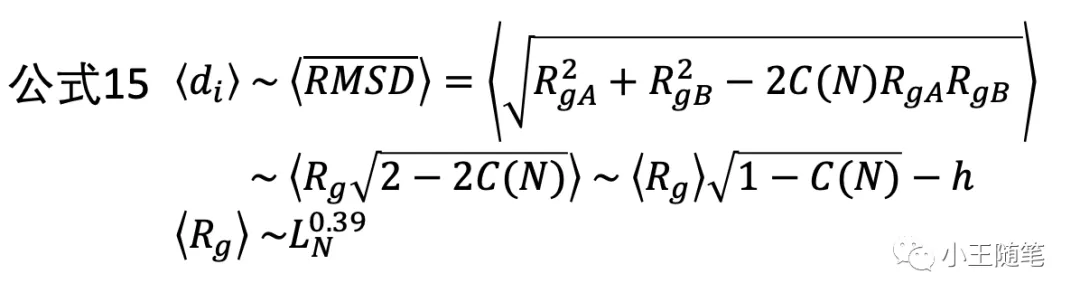
现在我们清楚了 TM-score 的物理意义:
LG-score + rRMSD —> TM-score
也就是说,依然如 RMSD 一般对齐两个相比较的结构,只不过这两个结构未必含有相同个数的残基,也就是可以只对齐结构的一个子集;对齐后,依然考虑两两对齐的残基之间的距离,只不过将这个距离除以它的统计平均值,并且这个平均值是链长的函数。
那么,由此可以凭直觉猜测 TM-score 有3大优势(改进):
- 不依赖链长,具体原因我们上面分析了。
- 不依赖序列(sequence independent),不需要两个相比较的结构有同样数目的残基。
- 最大程度地对齐两个结构的子集 —— 可衡量两个结构的整体拓扑。因为必须打分必须要除以 LN,所以,需要尽可能找到两个结构的最大的相似子集。
6年后,2010年,针对上述的第3点,张阳与学生 XU Jinrui 考察了 TM-score 在判断蛋白质 fold 方面的意义,量化了 TM-score 打分的统计意义:TM-score = 0.5 意味着两个比较的结构“很可能”同属一个 fold。[8] 这篇文章可能是 TM-score 获得巨大关注并被广为应用的原因 —— 我把我自己阐释清楚,从而别人也明白我。己知,而后人知。
一、前言
典型的基于比较建模/基于线程的蛋白质结构预测程序包括两个步骤:
- 查找与目标序列(即模板)相关的已知结构
- 基于模板构建全长模型
通常通过模型中等效原子与本构结构之间的均方根均方根偏差(RMSD)来评估所得全长模型的质量。但是,仅RMSD不足以估计初始模板的质量,因为对齐覆盖率在不同方法中可能会非常不同。显然,具有2ÅRMSD的模板到本地具有50%对齐覆盖率的模板并不能对于结构建模而言,它肯定比RMSD为3 Å但对准覆盖率为80%的模型更好。虽然模板对齐区域在前者中较好,因为对齐的残基较少,但生成的全长模型的质量可能较差。在开发有效的折叠识别算法期间,模板评估问题变得尤为重要,因为不同的序列-结构比对方案或参数可能会导致不同程度的比对置信度,并伴随着比对损失或增益。因此,一个单一的评估分数必须具有适当的对准精度(accuracy)和覆盖(coverage)范围之间的平衡,并且与最终全长模型的质量密切相关。同样重要的是,它必须区分随机预测和统计上重要的预测。
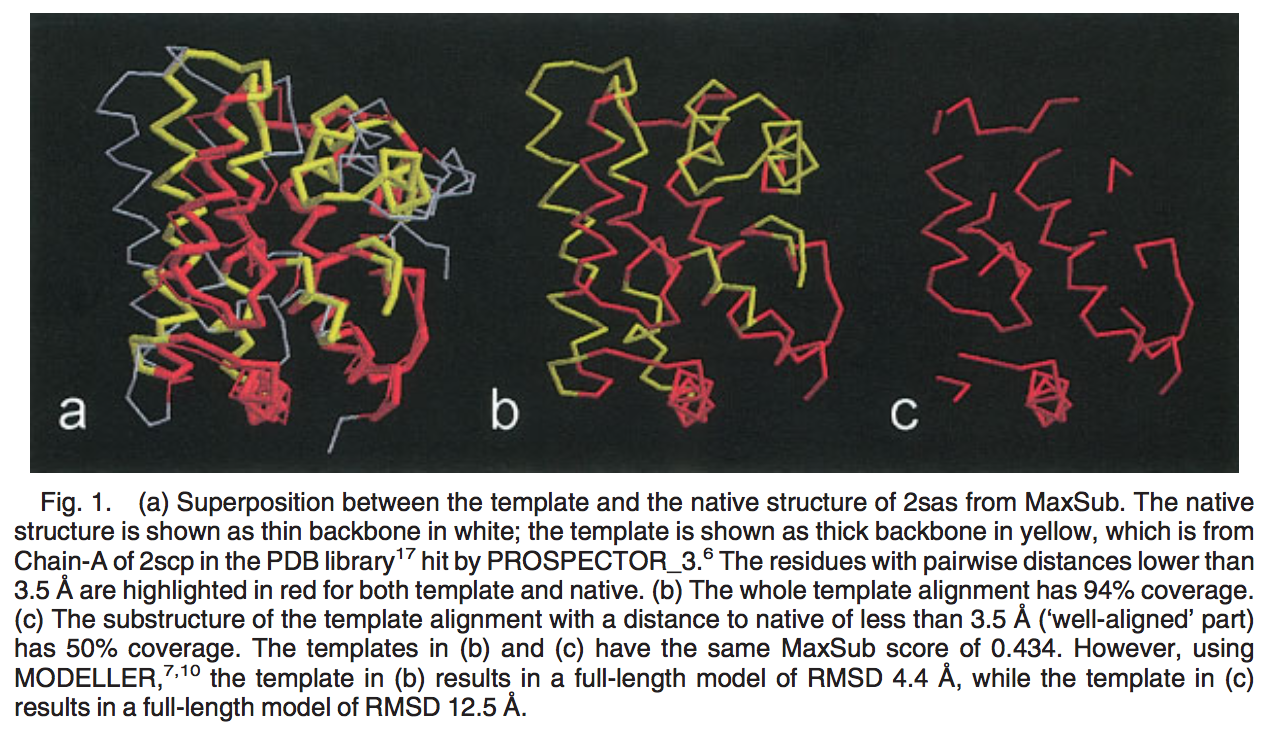
与上述问题高度相关,已经开发了一些有趣的评分功能,用于不同长度的两种结构的序列相关比较(与序列无关的结构比对算法相比)。例如,使用MaxSub, Siew和他的同事试图确定最大的子结构,其中两个结构叠加后的等效残基之间的距离小于某个阈值,例如3.5Å。由于MaxSub评分功能仅计算子结构中包含的那些残基,因此省略了子结构外部模板的空间信息。 例如,图1(a)显示了2sas_原始结构的MaxSub叠加和从线程程序PROSPECTOR_3获得的模板比对(94%的覆盖率,这是比对的残基数与靶标残留物数的比值),距离为3.5Å的残基对以红色突出显示(覆盖率50%),其余对齐的残基以黄色突出显示。 因此,图1(b)(原始对齐方式具有94%的覆盖率)和图1(c)(“对齐良好”的部分具有50%覆盖率)中的模板具有相同的MaxSub分数,该分数仅与集合相关红色残留物。但是,用于最终全长结构建模的模板的功能可能会显着不同。 例如,使用结构构建程序“模型”(MODELLER),图1(b)中的模板将生成一个全长模型,其RMSD来自本机为4.4Å, 而图1(c)中的模板产生的全长模型的RMSD值为12.5Å。通过进一步的说明,在PROSPECTOR_3比对的大型基准测试集中(见下文),有81个案例的MaxSub得分在0.4到0.45之间。由MODELLER建立的最终全长型号的RMSD值在3.5至35.7Å之间变化,标准偏差为4.8Å。因此,Maxsub得分与所得全长模型的质量之间没有明显的相关性。
在他们的GDT_TS评分功能中,Zemla和同事进一步确定了与几个不同阈值截止值相关的多个最大子结构(例如,最近的CASP5实验中使用的1、2、4和8 A)。 GDT_TS分数定义为具有四个不同距离阈值的子结构目标序列的平均覆盖率。 由于GDT_TS分数仅关注子结构的大小,因此模板/模型和本机结构的详细匹配信息会被部分遗漏(例如,与本机的偏差范围为4.1– 8AÂ的残基对评分功能的贡献相同 )。Zemla通过引入更多距离阈值进一步解决了这个问题。
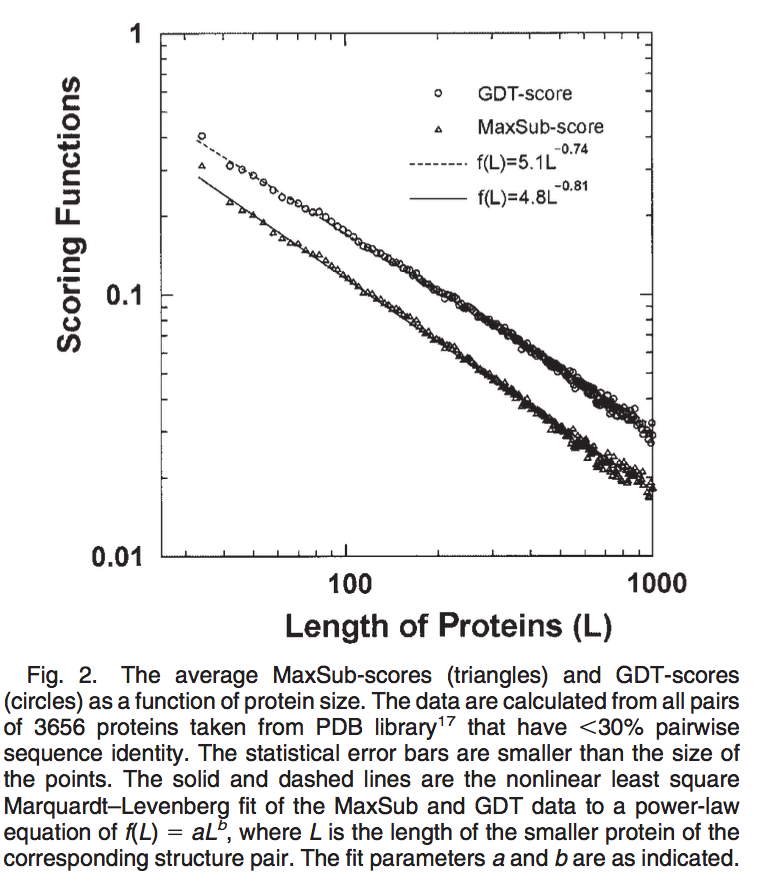
与这些评分功能相关的另一个问题是评分大小对所评估蛋白质大小的依赖性。 换句话说,必须解决以下问题:一对随机相关结构的对应得分值将是多少。 在图2中,我们绘制了蛋白质数据库(PDB)中成对序列同一性小于30%的随机结构对的平均MaxSub和GDT得分与蛋白质长度的关系。 这些分数显示了蛋白质大小的幂律依赖性。 显然,给定的绝对得分(例如GDT 0.4或MaxSub 0.3)可以反映出针对400个残基的目标的显着比对,但它接近于PDB中针对40个残基的目标的随机选择。 这种明显的大小依赖性使得这些评分函数的绝对量级变得毫无意义。
当通过结构比对或RMSD计算来测量结构相似性时,许多作者还观察到了随机相关结构对对结构相似性的显着蛋白质大小依赖性。 为了消除对蛋白质大小的依赖性,Levitt和Gerstein和Ortiz及其同事根据其随机结构数据库的统计信息,将其结构比对得分转换为统计显着性得分,称为P值。 对于他们的相对RMSD,Betancourt和Skolnick用大小和回转半径相似的随机结构对中的平均RMSD对RMSD进行归一化。 在RMSD-100评分中,Carugo和Pongor将RMSD除以 $ 1+ \sqrt{N/100}$的因子,其中N代表蛋白质长度。
在本文中,我们扩展了上述方法,并开发了用于评估线程(threading)模板的新评分功能,我们将其称为模板建模(TM)得分。我们的目的之一是重新缩放结构建模错误,以便分数值与随机相关结构对的蛋白质大小无关。由于线程模板最重要的用途之一是简化最终结构建模,因此我们的第二个目标是使初始模板的分数与最终全长模型的质量紧密相关。当然,正如许多作者所指出的那样,RMSD并不是完整模型质量的完美指标。除了随机结构对的显着大小依赖性外,当模型的其他部分具有较大的预测误差时,RMSD无法识别预测良好的子结构。在文学中,还有许多其他测量蛋白质建模质量的方法。以MAMMOTH为例,Ortiz及其同事通过比较本地和全局相似性来评估结构。在此,作为许多可能的选择之一,我们使用相对于RMSD(rRMSD)的Z分数表示来对最终全长模型的质量进行评分。我们考虑了一个大分数的基准蛋白质组,该序列涵盖了所有少于200个残基的蛋白质的35%序列同一性的当前PDB。初始模板的TM得分以及Maxsub和GDT_TS分数是根据它们与广泛使用的蛋白质建模建立的基准目标中最终全长模型质量的相关性进行评估的软件模型。
二、材料和方法
2.1 评分功能
我们的评分功能是Levitt–Gerstein(LG)评分的一种变体,该分数最初用于与序列无关的结构比对:
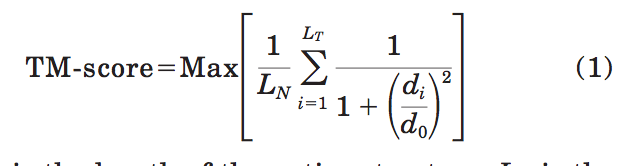
其中LN是自然结构的长度,LT是对齐的残基到模板结构的长度,di是第i对对齐的残基之间的距离,d0是将匹配差异归一化的标度。 “最大值”表示最佳空间叠加后的最大值。 TM分数的值始终介于(0,1]之间,更好的模板具有较高的TM分数。MaxSub中也使用了类似的公式,但求和仅限于那些具有di< d0的残基。 在LiveBench中,Rychlewski和同事定义了一个3维分数,其功能与LG分数相似,但格式与LG-score不同;在S-score中,Cristobal和同事使用 未归一的LG得分,包括空位罚分。如下所示,模板比对中的缺口数与最终模型的质量无关。
在所有上述方法中,d0的值都是恒定的。 例如,MaxSub中的d0为3.5Å,S-SCORE和原始LG分数为d0= 5Å。如图2所示,这些处理导致分数对随机蛋白质对中蛋白质大小的幂律依赖性。 在图3中,我们从PDB计算出具有成对序列同一性<30%的3656个蛋白质结构的平均TM得分。 如果取d0=5Å,我们称之为“原始TM得分”(rTM得分),我们发现它对蛋白质大小的幂律依赖性与MaxSub和GDT得分相似。
为了排除rTM评分中蛋白质大小的依赖性,我们首先对随机相关结构的平均结构匹配差异进行近似估算。 通常,LN长度相同的两个结构(A和B)的RMSD可以写成
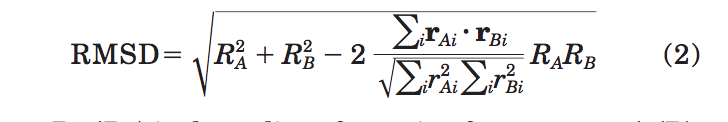
其中RA(RB)是结构A(B)的回转半径,rAi(rBi)是全局叠加后的坐标矢量。 通过对大约1300个非同源PDB结构的计算,Betancourt和Skolnick观察到,随机相关结构对的平均相关系数如下:

请记住,球状蛋白质结构的平均回转半径(verage radius of gyration of globular)与长度有幂律相关性(即
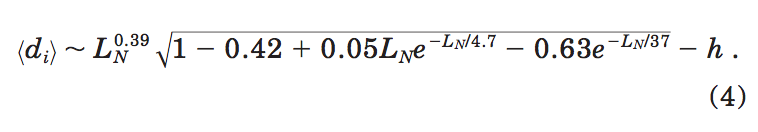
在这里引入常数h是因为TM分数计算中最佳局部结构匹配的平均距离始终小于RMSD计算中全局匹配的平均距离(请参见下文)。 当h =0.75时,可以通过一个简单的公式很好地近似eq(4)

例如,当LN从300个残基变为50个残基时,它会从6.4 A下降到2.3 A. 如图3所示,在eq(5)中定义的d0的TM得分具有大约为0.17的恒定值,与随机结构对的蛋白质大小无关。

2.2 搜索引擎
为了根据等式(1)找到模板的空间最优重叠和具有最大(或接近最大)TM分数的原始结构,我们使用了迭代搜索算法,类似于Zemla和 同事,Siew和同事,Ortiz和同事以及Kihara和Skolnick。
从由Lint相邻对齐残基组成的模板的初始片段开始,根据Kabsch的旋转矩阵,我们将该片段叠加到天然结构的相应残基上。 然后,我们收集到模板的所有残基,其与天然残基的距离小于d0,并将这组残基再次叠加到天然结构上。 重复该过程,直到旋转矩阵收敛为止。
由于收敛的叠加通常对片段Lint的初始选择敏感,因此我们分别使用Lint = LT,LT / 2,LT / 4,L,4进行了迭代过程。 当使用Lint <LT时,我们进行了所有迭代,初始片段的位置从N到C连续移动。 选择具有最高TM得分的旋转矩阵。
作为优化过程的确认,我们为上述1489个目标运行了搜索引擎约三倍的时间,并额外随机选择了初始片段。 只有92(6%)个案例的TM分数不同,所有差异均小于0.002。 基于旋转矩阵的高度收敛性,我们感到非常安全,可以得出结论,我们的搜索引擎对于得分最大化是最优的或接近最优的。
2.3 相对RMSD的Z得分
考虑到等式(2和3)中介绍的RMSD的回转半径和大小,Betancourt和Skolnick定义了一个相对RMSD(rRMSD)以消除依赖性:
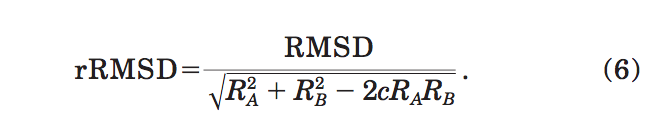
这只是感兴趣结构的RMSD值与相同回转半径的一对随机相关结构的平均RMSD之比。 随机结构对的rRMSD的标准偏差为
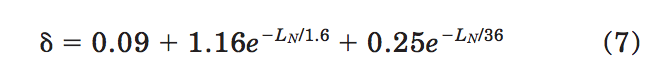
在我们的计算中,将rRMSD与平均值(其值为1)之类的Z分数定义为
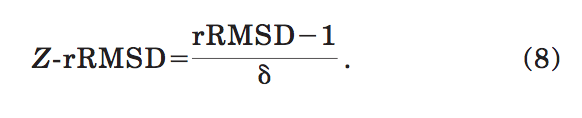
三、结果和讨论
3.1 目标和模板基准集
为了对评分功能进行可靠的评估,我们构建了一个全面的基准蛋白质组,其中包括1489个测试蛋白质,涵盖了PDB文库中其长度为41至200个残基,且35%的序列一致性。 1489种蛋白质的列表可在我们的网站上找到:http://www.bioinformatics.buffalo.edu/abinitio/1489 。 对于每个靶标,使用我们的线程程序PROSPECTOR_3获得模板结构,该程序旨在将靶标序列与从PDB选出的非同源可分辨结构文库进行匹配。与靶标的序列同一性为30%的模板蛋白为 从库中排除。 选择了PROSPECTOR_3比对中得分最高的模板。
3.2 模板的TM得分分布
在PROSPECTOR_3中,如果模板比对具有Z分数,则能量的标准偏差单位相对于平均值大于15,或者如果比对具有Z分数大于7,并且在结构上与相似Z-score的其他模板比对 ,通常对齐的可能性很高。 因此,根据PROSPECTOR_3,它被指定为“简易”目标,因为建模细化应比Z分数较低(通常具有较短的对准长度和较差的对准质量)的目标(称为“中等”)更容易 /硬目标。 在图4(a)中,我们显示了两种不同类别的线程模板的TM得分分布。 不出所料,大多数“简易”目标的TM得分都高于0.4,而大多数“中/硬”目标的TM得分都低于0.4,“简易”和“中/硬”之间存在明显差距 目标分布。
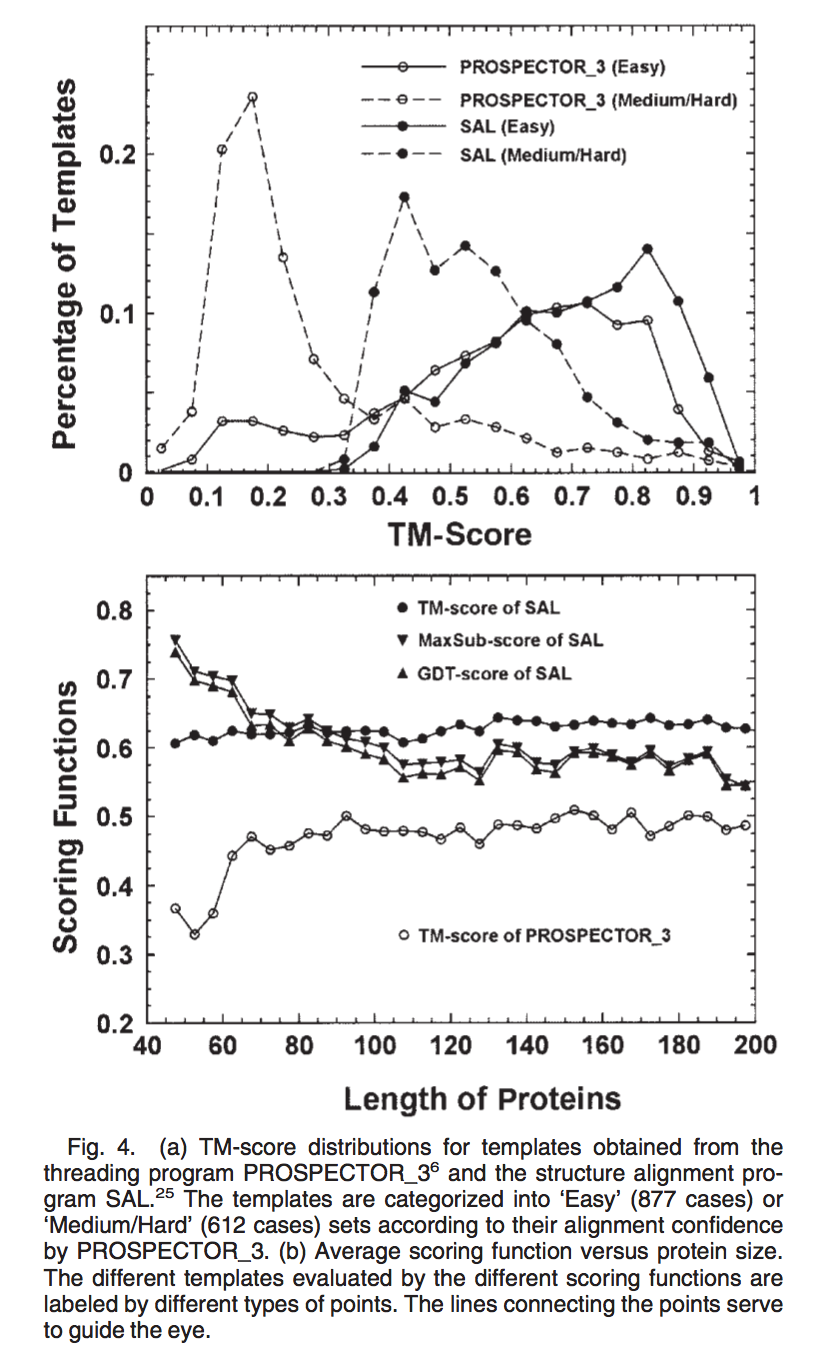
作为control,我们还在图4(a)中展示了从结构对齐程序SAL中识别出的模板的分布,该程序将本地结构与PROSPECTOR_3相同的模板库进行结构对齐,并返回带有如eq(8)中所定义的,最高的Z-rRMSD得分为原生。在“简易”组中,“黄金标准”结构比对具有约17%的序列同一性,PROSPECTOR_3的TM得分分布与SAL比对非常相似,除了PROSPECTOR_3的尾巴区域略有偏离TM分数小于0.4的实例更多。对于“中等/困难”情况,“金标准”结构比对具有约9%的序列同一性,远低于序列同一性的“暮光区”,大多数情况下,当前版本的PROSPECTOR_3失败:案件的TM分数小于0.4; 32%的情况下TM得分小于0.17,这意味着线程对齐比针对这些目标的随机选择没有提供更多的信息。当然,此结果与PROSPECTOR_3的评分系统一致,因为类别是根据其对齐可信度定义的。
真实线程(real threading)和“金标准”结构比对之间的比对差异也体现在它们对目标蛋白质大小的敏感性上。 如图4(b)所示,如eq(5)所示,在引入了尺寸依赖性标度后,SAL对齐的TM得分几乎没有尺寸依赖性。 另一方面,PROSPECTOR_3比对小目标的TM得分明显低于大目标,这突出了当前比对方法处理小蛋白的难度。 另一方面,由于对匹配差异使用了恒定的截止/比例,因此当通过MaxSub或GDT评分功能进行评估时,SAL对齐显示出明显的尺寸依赖性[见图4(b)]。
3.3 评分函数与最终全长模型之间的相关性
为了评估评分功能与从初始模板构建全长模型的能力之间的关系,我们使用PROSPECTOR_3模板作为唯一输入来构建全长模型。 为了使评估的通用性和明确性起见,我们采用了最广泛使用的建模软件程序之一,MODELLER,该程序旨在通过最佳地满足从输入模板中提取的空间约束来构建全长模型。 该算法代表了不同作者开发的一套通用方法。
图5(a–c)分别显示了线程模板的TM得分,MaxSub得分和GDT_TS得分与MODELLER构建的最终全长模型的Z-rRMSD的关系。 在这里,我们仅介绍MODELLER可以为其生成Z-rRMSD对本机的模型低于1的模型的那些目标(1489中的1048),因为对于太弱相似性的结构对,评分评估变得不那么重要。 与其他两个评分函数和最终模型中的任何一个之间,TM得分与最终全长模型的质量之间存在明显更强的相关性。 更准确地说,我们将相关系数定义为


其中S和Z分别代表初始模板的评分函数,以及Z-rRMSD到最终全长模型的本征函数。 <…>的平均值仅包括那些最终模型的Z-rRMSD值小于1的目标。根据图5(a–c),TM得分与Z-rRMSD的相关系数最高(0.891)。 在所有三个得分函数中的最终模型,GDT_TS得分(0.751)略好于MaxSub得分(0.746)。
在图5(d)中,我们将Z-rRMSD空间划分为20个bin,并为每个Z-rRMSD bin计算初始模板的评分函数的波动。 通常,模板评分的标准偏差会随着最终模型的Z-rRMSD值而增加。 与相关系数一致,对于三个分数的给定Z-rRMSD值,TM分数具有最小的色散。 对于非常好的Z-rRMSD模型,GDT分数和MaxSub分数的色散相似。 当Z-rRMSD的绝对值减小(或模板的质量通常变差)时,与MaxSub得分相比,GDT得分由于其多个阈值截止分辨率而更能代表最终模型 质量。 从大于5.3的Z-rRMSD开始,某些模板的MaxSub得分为0,这表明这些目标对最终模型的质量不敏感。
在所有1489个目标中,有50%的线程模板的TM得分高于0.48。如果我们将成功预测的最终模型视为Z-rRMSD值小于5的模型,则TM得分大于0.48的模板的假阳性率为16.5%,而TM-得分的模板的假阴性率为分数小于0.48是4.1%。同样,50%的目标具有MaxSub-scores高于0.42的线程模板。使用相同的最大Z-rRMSD(5)作为成功预测的阈值时,MaxSub得分的误报率和误报率分别为23.5%和9.2%。对于GDT分数,有50%的目标具有GDT分数大于0.475的线程模板,且误报率和误报率分别为23.0%和7.9%。因此,基于其选择质量较好,假阳性和假阴性最少的模型的能力,TM得分最高,Maxsub和GDT得分表现出基本相同的性能。
3.4 模板比对的间隙密度 Gap Density of Template Alignments
TM得分与原始LG-和S-score公式之间的区别之一是,后者得分中包含-10 X Ngap的空位罚分,其中Ngap是对齐空位的数量。 包含空位罚分可能导致某些比对中的得分为负。
为了进一步检查TM分数,我们检查了模板比对中发现的缺口数对最终建模结果的平均影响。 我们将归一化的缺口数(或缺口密度)定义为Ngap与对齐残基数的比率,其中Ngap计算为Nfra - 1。Nfra表示由两个以上残基组成的连续碎片数。 数据显示,间隙密度与最终模型的Z-rRMSD之间基本上没有相关性(相关系数为0.004)。
我们还研究了将空位罚分组合为等式的不同方法。 (1); 没有一个可以改善最终全长模型的TM得分与Z-rRMSD之间的相关性。 原因可能是在包括MODELLER在内的大多数模型构建程序中,最终模型的质量取决于从模板提取的第三级空间约束以提供全局折叠信息。 这样的信息对比对中的缺口数目不敏感。
3.5 蛋白质结构预测的自动评估
如最近的CASP实验(对结构预测的关键评估)所示,对预测的三级模型进行准确而自动的评估并非易事,因为在质量较低的模型中,不同的度量标准对不同的特征敏感。 在CASP5中,评估人员开发了各种综合评分功能,可有效地用于比较模型的自动评估和对折识别目标的预测。但是,对于困难目标,特别是那些“新折”目标,通常需要进行人的视觉评估。
尽管开发TM分数的主要目的是评估部分对齐的模板与最终的最终全长模型之间的关系,但我们也将根据完整质量对TM分数进行评估 长模型,尤其适用于hard New Fold targets。 由于这些模型与实验结构的相似性较弱,并且缺乏生物学洞察力,因此在这里,我们将CASP5中的专家经验评估作为“黄金标准”。
在图6中,我们对从五个新的折叠目标的不同组提交的第一个预测模型进行排名,这些目标分别是从CASP5网页下载的T0129,T0149_2(203-318),T0161,T0162_3(114-281)和T0181: http://predictioncenter.llnl.gov/casp5/pubResultS/CASP_BROWSER ,并根据TM得分(第一列),MaxSub得分(第二列)和GDT得分(第三列)计算这些模型的得分函数)。根据Aloy和同事的人类视觉评估(数据来自 http://www.russell.embl.de/casp5/NF/Table2_1st.html ),模型在视觉时获得2分(“优秀”)评估发现总体折叠是正确的,或者当模型被认为是正确折叠的中间部分时得分为1分(“好”),其他所有模型的得分均为0。在五个新折叠目标的所有提交模型中,在Aloy和同事的评估中,有3个模型的得分为“优秀”,而有7个模型的得分为“良好”。这些模型在图6中以实心正方形(优秀)和实心圆圈(良好)突出显示。
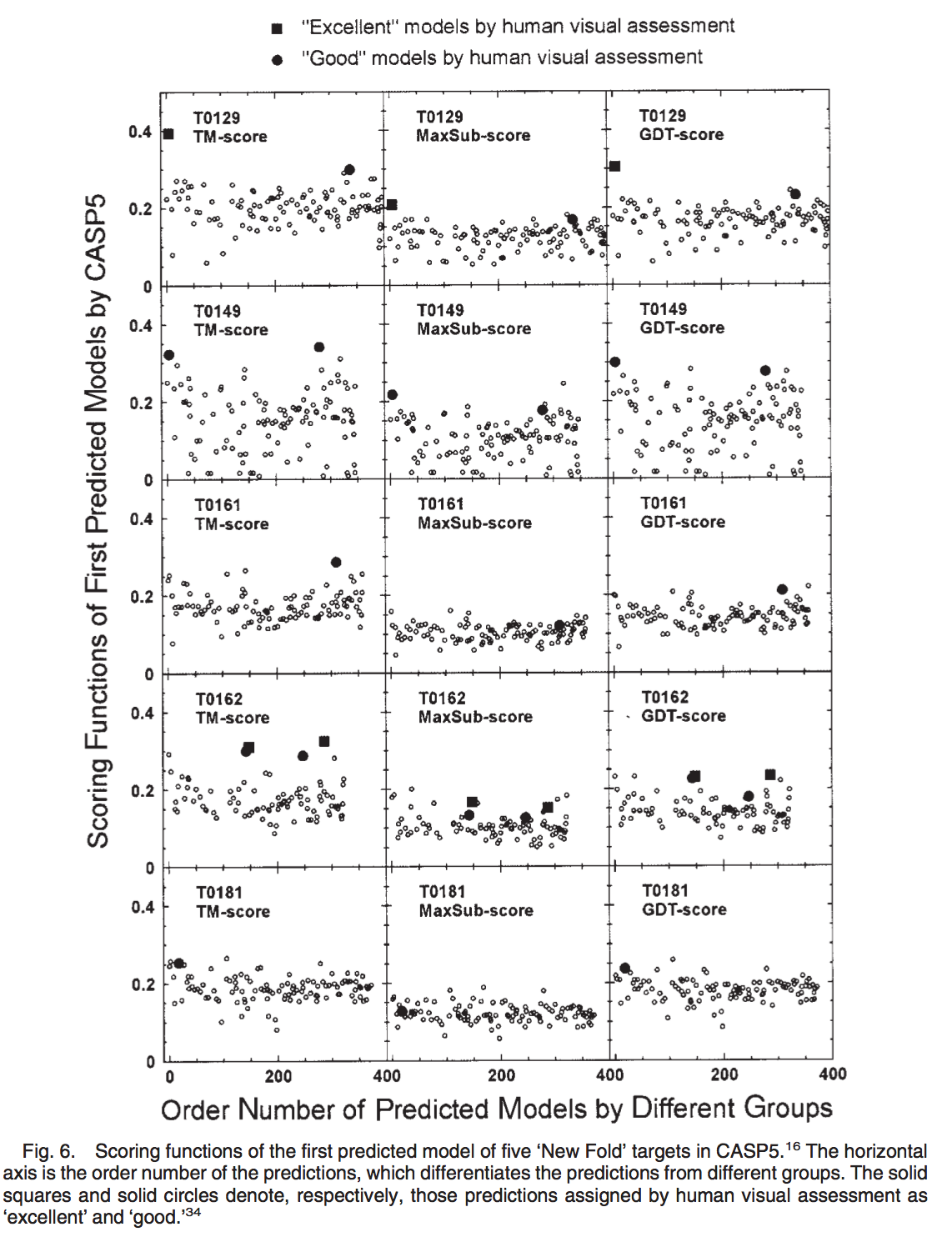
如图6的第I列所示,根据人类视觉评估,几乎所有这些成功预测的模型的TM得分都明显高于人类专家认为不正确的其他预测。 标记为“优秀”的模型的TM得分也比标记为“良好”的模型的得分高一些。通常,使用TM得分或GDT得分可以将优良/优良模型与错误模型区分开; 使用MaxSub-score时不是这种情况。 但是,仍然存在一些示例,例如T0162,其良好模型的GDT分数低于许多错误模型的GDT分数(请参见图6中第4行第III列)。
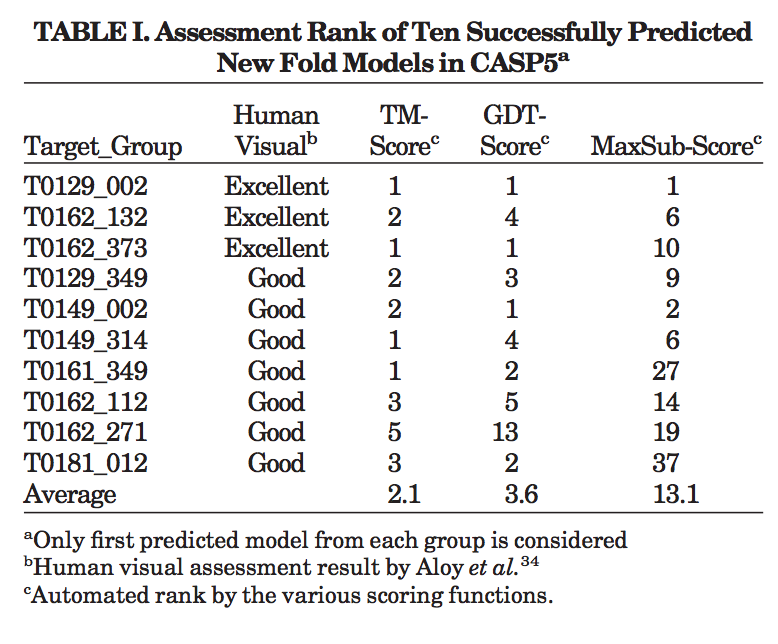
表I分别根据TM得分,GDT得分和MaxSub得分列出了所有十个成功预测模型的自动排名。 与图6中的数据一致,TM得分评估的平均排名(2.1)比GDT得分(3.6)略好; 两者均比MaxSub分数评估的平均排名(13.1)好得多。
四、结论
我们已经开发了一种新的评分功能,可以自动评估蛋白质结构预测。
- 首先,通过蛋白质大小依赖性尺度对建模误差进行归一化,以使随机蛋白质对的平均TM得分对目标蛋白质的长度没有偏倚,从而为线程对齐或最终模型预测任何有意义的设置了最小阈值,即TM得分0.17。
- 其次,不是像MaxSub或GDT评分功能那样使用特定的距离截止并只关注结构的分数,而是在TM评分中评估了建模蛋白质的所有残基。
为了进行客观评估,我们从线程程序PROSPECTOR_3中构建了蛋白质大规模基准测试集的结构模板,并使用广泛使用的程序MODELLER构建了全长模型。我们选择MODELLER的原因是它的方法学构成了许多比较建模工具的基础,并且被结构生物学家广泛使用。根据建模结果,TM评分与最终全长模型的质量之间的相关性比MaxSub- score2或GDT_TS评分强。我们还从基准组中随机选择了200种蛋白质,并使用不同的建模算法TASSER对其进行了重建,该算法旨在通过重新排列初始模板比对中的连续片段来组装全长模型,并具有完善比对的能力。残基使其更接近天然结构。结果在质量上与此处使用MODELLER获得的结果相似,尽管在TASSER改进中,所有三个得分的平均相关性均较弱。 (这是因为TASSER生成的平均RMSD较低,而平均生成的RMSD最终模型更好,而平均本机则较低。)这表明TM分数描述的模板的功能对特定的优化方法不敏感。显然,与任何实际的全长建模优化程序相比,TM得分可更快,更方便地用于模板质量的判断。
TM分数显示的初始模板比对与最终模型之间的相关性比MaxSub分数更紧密的原因是,TM分数对高精度对齐区域和低精度对齐区域的模板信息进行计数,而 MaxSub分数忽略了低精度对齐区域中包含的对齐信息,该信息可能有助于全局建模。 另一方面,与RMSD的所有残差均以相等的权重对预测误差进行平均不同,TM得分使用LG因子,该因子分别对低和高精度区域进行加权。 这也允许TM得分比GDT得分提供更敏感的度量。 当使用TM来识别良好的模板比对时,与Maxsub或GDT得分相比,全分子组装的假阳性率和阴性率更低,从而验证了这一评估。
我们进一步利用TM得分来评估CASP5中五个可用的“ New Fold”目标的预测全长模型。第一个预测模型的TM得分等级与Aloy和同事的专家视觉分析得出的等级非常吻合。。 通过TM得分,所有十个成功预测模型的平均排名为2.1(GDT得分为3.6,MaxSub得分为13.1)。 该结果表明,TM得分也可以用作蛋白质全长结构预测的自动评估的有用补充。
可从 http://bioinformatics.buffalo.edu/TM-score 免费下载TM-score程序。 为了方便用户,该程序还基于与TM分数相同的搜索引擎,提供了MaxSub分数和GDT_TS分数输出的选项。 对于大约200个残留物的结构,在2.4 GHz Pentium-4处理器上的计算时间不到1秒。
参考资料
- https://zhanglab.ccmb.med.umich.edu/TM-score/TM-score.pdf 。 Proteins 2004;57:702–710. © 2004 Wiley-Liss, Inc.. Yang Zhang and Jeffrey Skolnick
- [5] Zhang & Skolnick, Scoring function for automated assessment of protein structure template quality, Proteins (2004)
- [6] Levitt & Gerstein, A unified statistical framework for sequence comparison and structure comparison, PNAS (1998)
- [7] Siew et al., MaxSub: an automated measure for the assessment of protein structure prediction quality (2000)
- [8] Xu & Zhang, How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics (2010)
