基因集富集分析(GSEA)及其可视化
需要阅读:
- 更多有意思的结果展示,参考: https://www.jianshu.com/p/3c5de0c4ca76
- https://www.pathwaycommons.org/guide/primers/data_analysis/gsea/ (解读很到位)
传统KEGG(通路富集分析)和GO(功能富集)分析时,如果富集到的同一通路下,既有上调差异基因,也有下调差异基因,那么这条通路总体的表现形式究竟是怎样?是被抑制还是激活?或者更直观点说,这条通路下的基因表达水平在实验处理后是上升了呢,还是下降了呢?
传统的富集分析,针对总体的差异基因,不区分哪些差异基因是上调还是下调。
这是因为,传统的富集分析根本不考虑基因表达量的变化趋势,其算法的核心只关注这些差异基因的分布是否和随机抽样得到的分布一致,即使后期绘图时,在通路图上用不同颜色标记了上下调的基因,但,由于没有采用有效的统计学手段去分析某条通路下的差异基因的总体变化趋势,这使得传统的富集分析结果无法回答上述的问题。
于是,有人提出,分别提取上调或者下调的差异基因来进行传统富集分析,由于事先根据上调下调(表达量变化趋势)对差异基因进行了筛选,从而回避了上面的问题。但,这样的做法有失偏颇,因为费舍尔精确检验就是想要证明这个差异基因列表不是随机抽样得到的,而事先对差异基因列表的进行上调或下调的过滤,就会对结果的随机性造成了干扰,最后得出的结论其准确性也受影响。而且,在细胞内发挥生物学功能时,上调和下调基因是共同发挥作用,进行富集分析时,将上下调分开进行分析,也不符合实际情况。
想象一下,上调基因和下调基因分开富集,然后富集到了同一条通路,这怎么解释?
所以,准确来说,传统的富集分析只能定位到功能,这些差异基因与哪些功能相关,而不能回答某条通路的整体表现形式。想要回答这个问题,所以我们需要GSEA富集方法的结果。
一、 什么是GSEA
GSEA(Gene Set Enrichment Analysis):基因集富集分析,由Broad Institute研究所提出的一种富集方法,同时还提供对应的分析软件GSEA和一个基因集数据库MSigdb[http://software.broadinstitute.org/gsea/msigdb/index.jsp]

GSEA的原理图
基因集富集分析(Gene Set Enrichment Analysis, GSEA)是是一种计算方法,用于确定事先定义的一组基因是否在不同的样品中差异表达。
GSEA首先将基因在样品中的差异倍数值(logFC)由大到小排序,然后判断来自功能注释等预定义的基因集或自定义的基因集中的基因是富集在这个排序列表的顶部还是底部,如果在富集顶部,则该基因集是上调趋势,反之,如果富集在底部,则是下调趋势。
GSEA特点
GSEA是个非常强大的富集分析方法,可以针对多种数据库中的数据进行GSEA分析,包括常见的GO数据库,KEGG数据库,Reactome数据库以及MSigDb数据库或其他自定义数据集。并且与传统的GO/KEGG只对显著的差异基因进行功能注释相比,GSEA
-
考虑了基因的表达水平,不需要指定明确的差异基因阈值,检验的是基因集合而非单个基因的表达变化,算法会根据实际数据的整体趋势进行分析。
-
GO和KEGG只能定位到功能,而GSEA通过检验预定义的基因集的基因在某种状态下表达水平而提示该通路是否被激活还是抑制。
-
以待测功能基因集为对象来进行检验,使得检验结果针对性和灵敏性提高。
分析逻辑
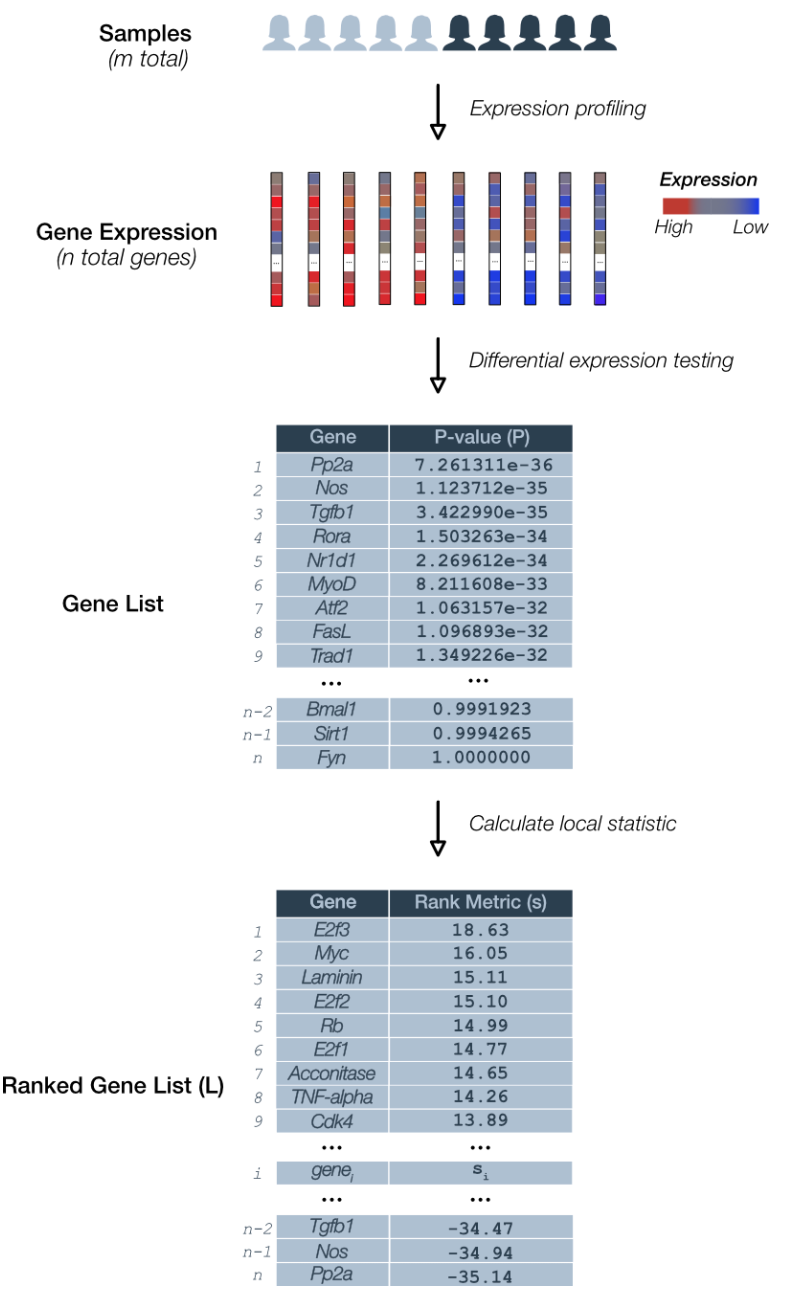
Figure 1. Deriving the GSEA local statistic: Rank metric. A pairwise comparison of gene expression for m total samples is depicted. RNA levels for each of n genes is determined. Differential expression testing assigns a p-value (P) to each gene and is used to derive the local statistic denoted the GSEA rank metric (s). A gene list (L) is ordered according to rank.

Figure 2. Sample calculation of global statistic: The GSEA enrichment score. The process requires the ranked gene list (L) ordered according to the ranking metric along with a candidate gene set (G). In this case, the candidate is a mammalian cell cycle pathway. A running sum is calculated by starting at the top of the ranked list and considering each gene in succession: Add to the sum if the gene is present in gene set (red; +) and decrement the sum otherwise (-). The GSEA enrichment score (S) is the maximum value of the sum at any point in the list. Although not shown, the running sum may deviate in the negative direction, hence, S is actually the largest absolute value of the running sum.

Figure 3. Empirical cumulative distribution functions. (Above) A hypothetical ordered gene list where each line represents a gene in the gene set (red) or not (blue). (Below) Empirical cumulative distribution functions for the genes in the gene set (red) and those outside the gene set (blue). The maximum deviation between ecdfs is the dotted green line.
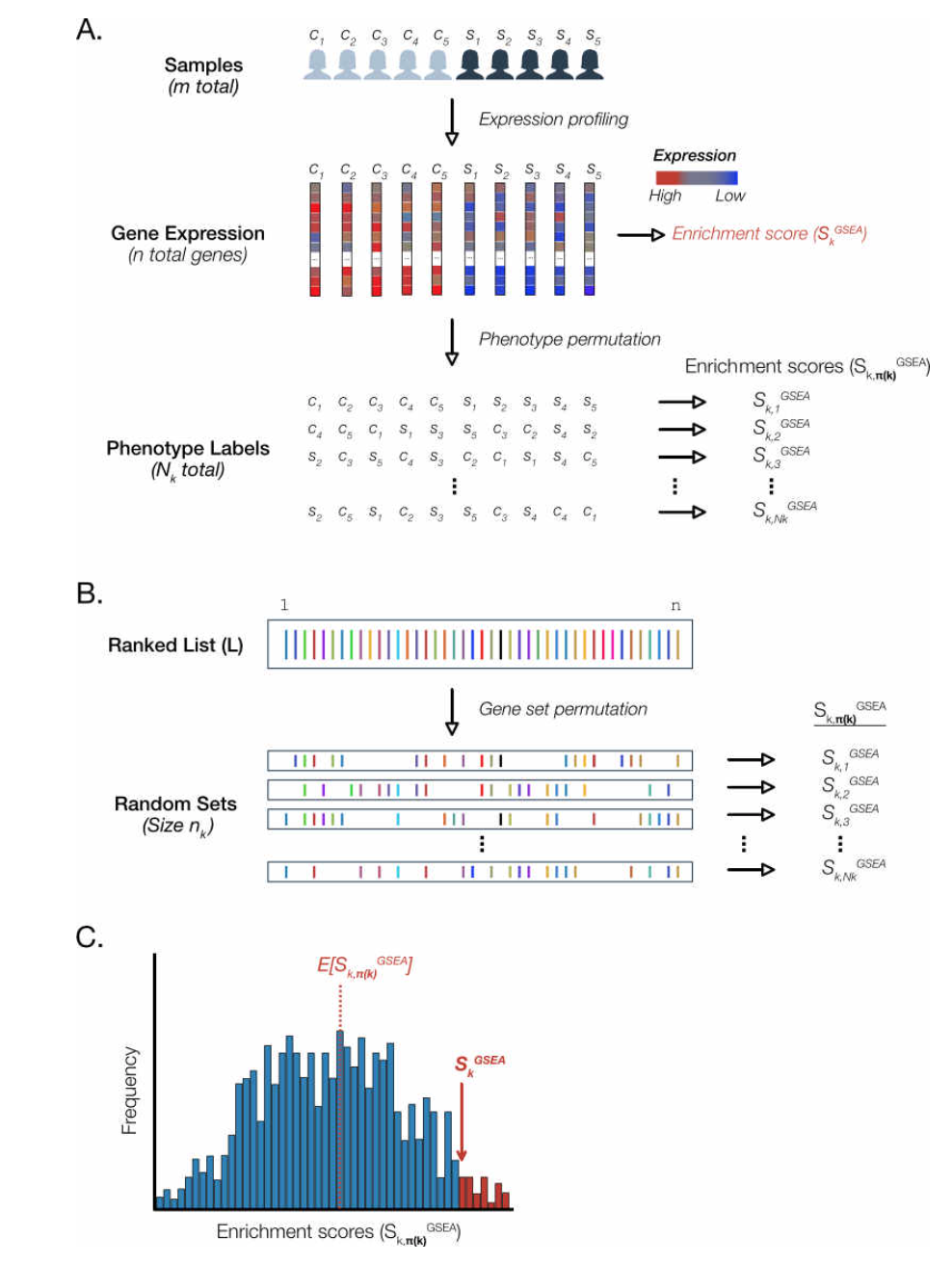
Figure 4. GSEA uses permutation methods to generate null distributions for each gene set. For the sake of brevity, we depict a schematic of permutation methods for a single gene set. In GSEA, this process is repeated separately for each gene set. A. Phenotype permutation. B. Gene set permutation. C. Calculation of p-values.
二、GSEA原理解读
GSEA分析的是一个基因集下的所有基因是富集在这个排序列表的顶部还是底部,如果在顶部富集,可以说,从总体上看,该基因集是上调趋势,反之,如果在底部富集,则是下调趋势。
GSEA分析有三个特点:
- 分析的基因集合而不是单个基因;
- 将基因与预定义的基因集进行比较;
- 富集分析;
假设1个比较组“MUT vs WT”的差异gene集(MUT为实验组,WT为对照组),进行GSEA富集分析,结果如下图:
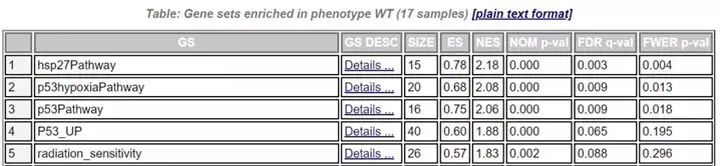
- GS:基因集(通路)的名字。
- SIZE:代表该基因集(通路)下的基因总数。
- ES:代表Enrichment score,NES代表归一化后的Enrichment score。
- NOM p-val:代表p值,表征富集结果的可信度。
- FDR q-val`代表q值, 是多重假设检验矫正后的p值,注意GSEA采用pvalue < 5%, qvalue < 25% 对结果进行过滤。
对于某个基因集下(通路里)的每个基因给出了详细的统计信息,如下图
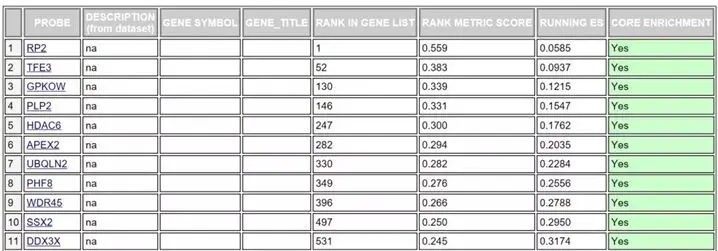
- RANK IN GENE LIST:代表该基因在排序中的位置。
- RANK METRIC SCORE:代表该基因排序量的值,即:处理后的foldchange值。
- RUNNIG ES:代表累计的Enrichment score。
- CORE ENRICHMENT:代表是否属于核心基因,即对该基因集的Enerchment score做出了主要贡献的基因。
上图表格中的数据对应下面这张图
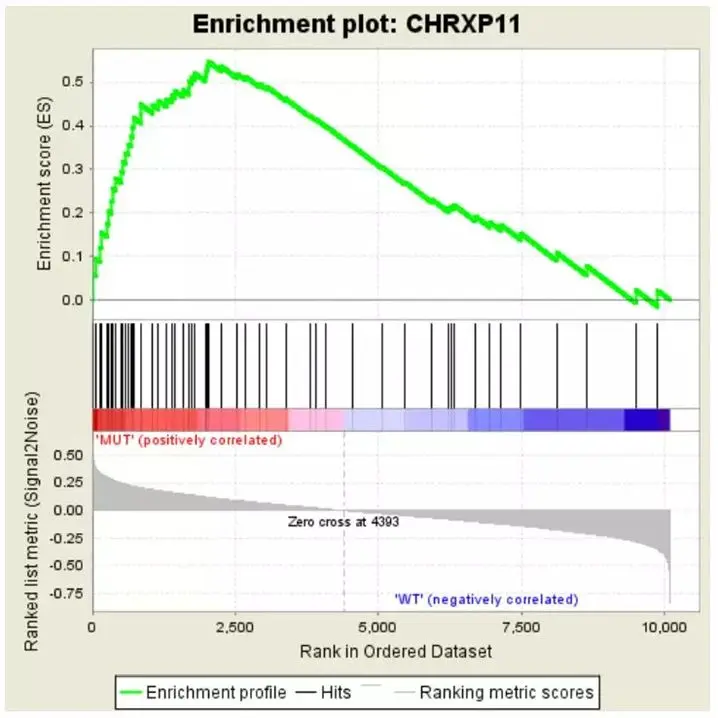
Enrichment Score的折线图
上图分为3部分,如下:
-
第一部分:最顶部的绿色折线为基因Enrichment Score的折线图。纵轴为对应的Running ES, 在折线图中有个峰值,该峰值就是这个基因集的Enrichemnt score,峰值之前的基因就是该基因集下的核心基因。横轴代表此基因集下的每个基因,对应第二部分类似条形码的竖线。
-
第二部分:类似条形码的部分,为Hits,每条竖线对应该基因集下的一个基因。
-
第三部分:为所有基因的rank值分布图,纵坐标为ranked list metric,即该基因排序量的值,可理解为“公式化处理后的foldchange值”。
我们假设是针对比较组“MUT vs WT”的差异gene集进行分析,MUT为实验组,WT为对照组,差异gene的差异倍数计算公式为:
foldchange = 实验组表达量/对照组表达量
通常统计时,对foldchange取log值(取对数)。如果log2(foldchange)>0,表明实验组表达量高于对照组,即,实验组相对于对照组上调。如果log2(foldchange)<0,表明实验组相对于对照组下调。 上图“Enrichment Score的折线图”对应的纵轴值全部大于0,显示这个基因集是在MUT组高表达的(即,此基因集(通路)在MUT组上调),下图是一个在WT组中高表达的示例。
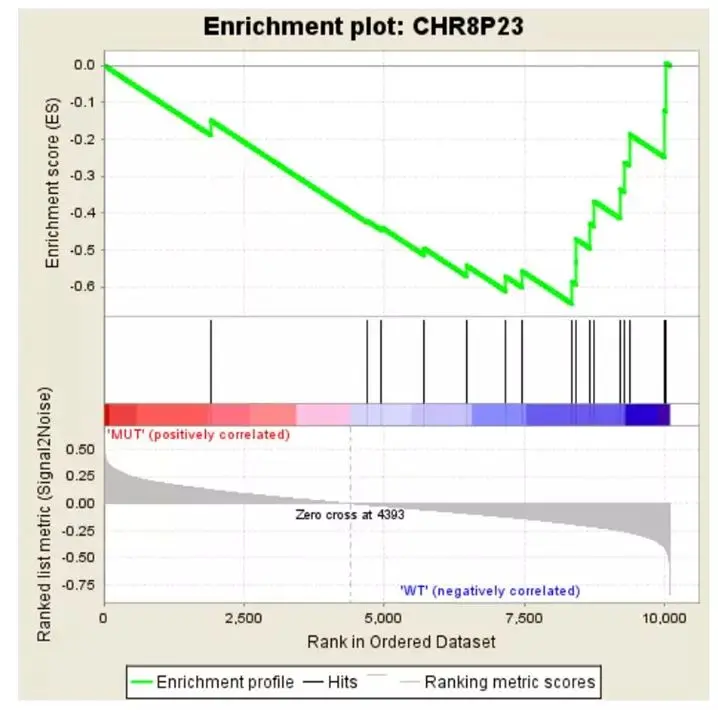
下图中其Enrichment score值全部为负数,其峰值右侧的基因为该基因集下的核心基因。
对于一个基因集而言,定义其中对Enrichment score贡献最大的基因为核心基因,也称之为leading edge subset, 参考下图
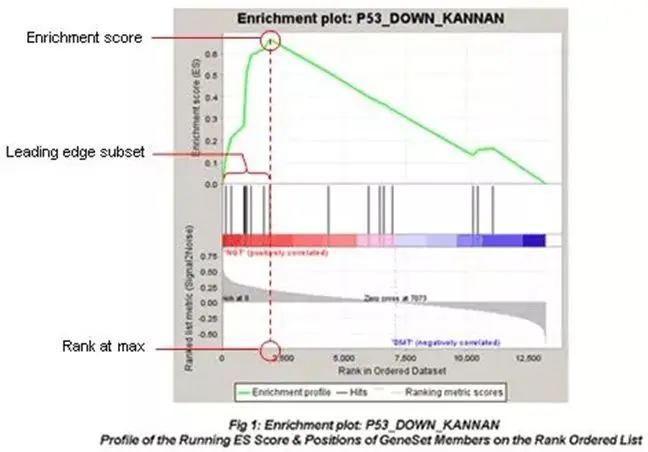
对于Enrichment score为正数的基因集而言,其核心基因是峰值之前的基因,对于Enrichment score为负数的基因集而言,其核心基因是峰值之后的基因。
上表KEGG富集分析结果,最后一列为LEADING EDGE, 在这一栏中,包含以下3个统计量:tags、list、signal tags表示核心基因在该基因集基因总数的占比,而list表示核心基因占所有基因总数的比例,signal利用这两个指标计算得到,公式如下
(Tag % )(1-Gene %)(N/(N-Nh))
N代表所有基因的数目,Nh代表该基因集下的基因总数。对于一个基因集而言,当核心基因的数目和该基因集下的基因总数相同,signal取值最大,当该基因集的基因数目和所有基因数目接近时,signal的取值接近于0。
GSEA软件默认的输入是基因集合(基因表达量矩阵和样本分组),然后内置的进行归一化,进行差异分析,计算singal2noise等统计量,其本质就是进行差异分析,计算出类似foldchange的统计值,但,GSEADE 归一化算法是否适用于我们输入的表达量矩阵,在计算基因的foldchange值时有没有考虑生物学重复本身的变化程度,这些都导致其计算出的foldchange值并不能满足我们的需求,更加有效的做法是采用专用的差异分析软件(比如DESeq2、Edger等)计算出的foldchange值来进行富集分析。
GSEA的核心是Enrichment score的计算,除了GSEA软件外,还有很多的工具也都支持这个算法,如果想要利用DESeq2等工具自定义计算出的基因排序列表进行富集分析,更推荐使用cluster Profiler等第三方工具。
GSEA富集过程包括三步骤:
- 计算富集分数(Enrichment Score);
- 估计富集分数的显著性水平;
- 矫正多重假设检验;
GSEA的具体原理请见PNAS文章:Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005 Oct 25;102(43):15545-50. Epub 2005 Sep 30.
小结
常规富集分析侧重于比较两组间的基因表达差异,集中关注少数几个显著上调或下调的基因,这种方式可能由于筛选参数的不合理导致漏掉一些关键信息,比如,部分差异表达不显著却有重要生物学意义的基因。因为实际通过测序检测到的RNA表达变化,是层层的负反馈调控后的结果,并且不同组织对于表达差异的敏感度是不同的:在神经递质系统内,一个1.2倍的表达差异即可能产生极其显著的效应。
下图示例中,无论设置怎样的差异筛选条件,如果仅做普通的Pathway富集分析的话,一定会漏检至关重要的Myc通路。这个示例非常典型,不仅在于Myc作为重要的癌基因广为人知,并且这里Myc在实验条件下活性改变后引起的下游基因表达变化,非常具有代表性:即并非所有的下游基因都会展现出强烈的表达改变,但它们会呈现出一致的趋势。GSEA的优势在于,无需做差异分析,直接用所有基因的表达量进行分析,可检测出不显著但是一致的差异表达趋势的基因集。
GSEA也可用于判断,某条通路在某组样本中是激活还是抑制。
三、 GSEA结果解读
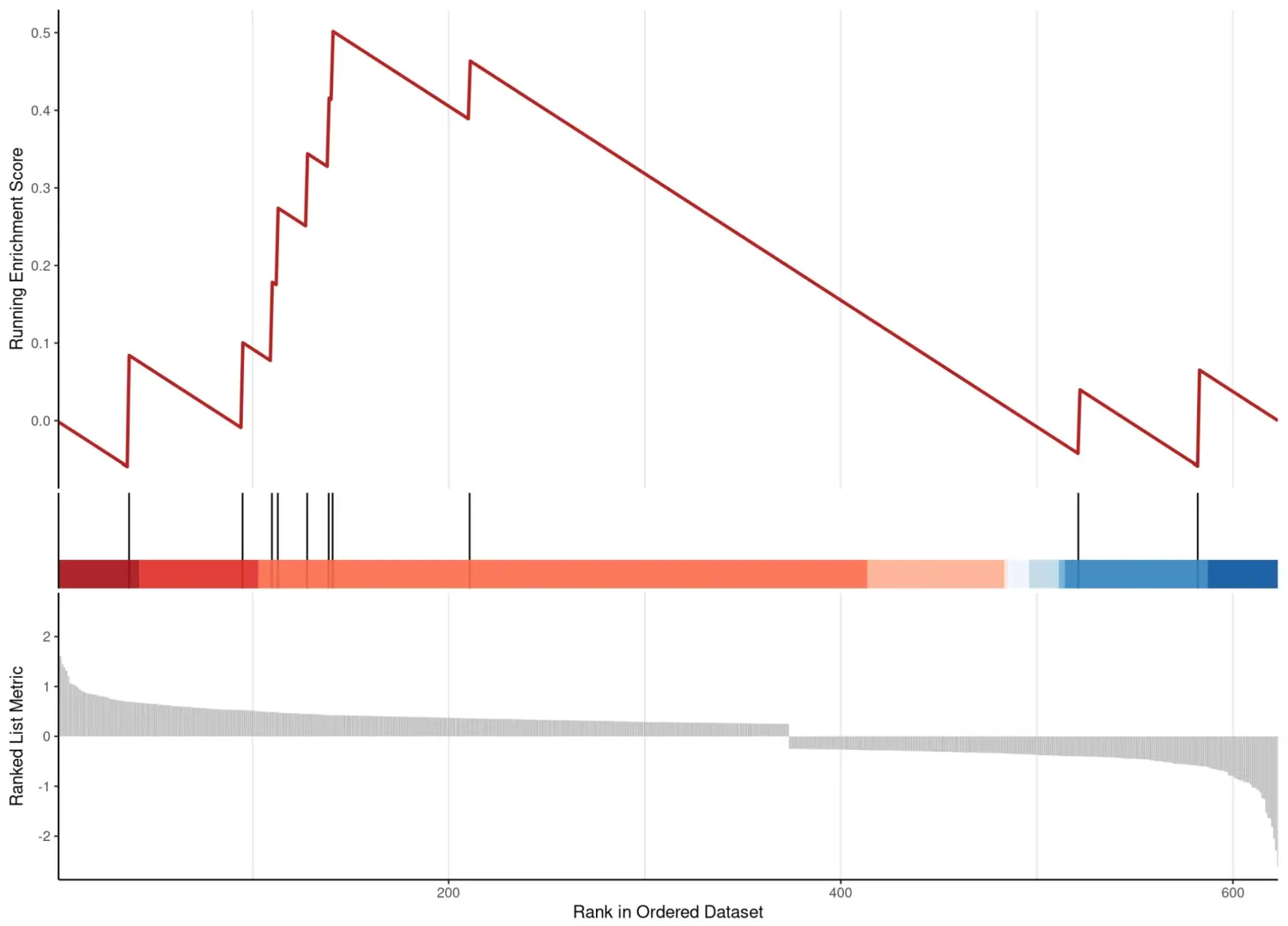
-
第1部分:Enrichment Score的折线图。 横轴为排序后的基因,纵轴为对应的ES, 在折线图中出现的峰值就是这个基因集的富集分数(Enrichment Score,ES)。ES是从排序后的表达基因集的第一个基因开始,如果排序后表达基因列表中的基因出现在功能基因数据集中则加分,反之则减分。正值说明在顶部富集,峰值左边的基因为核心基因,负值则相反。
-
第2部分:基因位置图。 黑线代表排序后表达基因列表中的基因位于当前分析的功能注释基因集的位置,红蓝相间的热图是表达丰度排列,红色越深的表示该位置的基因logFC越大 ,蓝色越深表示logFC越小。如果研究的功能注释基因集的成员显著聚集在表达数据集的顶部或底部,则说明功能基因数据集中的基因在数据集中高表达或低表达,若随机分配,则说明表达数据集与该通路无关。
-
第3部分:每个基因对应的信噪比(Signal2noise)。 以灰色面积图展示。灰色阴影的面积比,可以从整体上反映组间的Signal2noise的大小。
-
一般认为校正后的富集分数(NES)|NES|>1,p<0.05, qvalue(即FDR)<0.25的结果有意义。
四、GSEA实战
安装并导入要用到的R包
BiocManager::install("clusterProfiler") #感谢Y叔的clusterprofiler包
BiocManager::install("enrichplot") #画图需要
BiocManager::install("org.Hs.eg.db") #基因注释需要
library(org.Hs.eg.db)
library(clusterProfiler)
library(enrichplot)
导入数据
#library(readxl)
#setwd("D:/project/cancer/IL12/1.analysis/biomarker/")
#deg <- read.csv('pair-pre-post/differential_gene.txt',sep=' ')
#加载数据 数据链接:https://wwu.lanzouf.com/idmJB07bcefa
load(file = 'GSEA_test.Rdata')
colnames(deg)
head(deg)
#得到差异基因列表后取出 ,p值和logFC
nrDEG=deg[,c(2,1)]
colnames(nrDEG)=c('log2FoldChange','pvalue')
head(nrDEG)
log2FoldChange pvalue
LYZ -1.812078 1.228136e-144
FCGR3A 2.617707 3.977801e-119
S100A9 -2.286734 2.481257e-95
S100A8 -2.610696 3.626489e-92
IFITM2 1.445772 7.942512e-87
LGALS2 -2.049431 1.275856e-75
#注:最后需要为文件如上:一列为基因名,一列为FC值,一列为p值
#加载Y叔的R包,把SYMBOL转换为ENTREZID,后面可以直接做 KEGG 和 GO
library(org.Hs.eg.db)
library(clusterProfiler)
gene <- bitr(rownames(nrDEG), #转换的列是nrDEG的列名
fromType = "SYMBOL", #需要转换ID类型
toType = "ENTREZID", #转换成的ID类型
OrgDb = org.Hs.eg.db) #对应的物种,小鼠的是org.Mm.eg.db
#让基因名、ENTREZID、logFC对应起来
gene$logFC <- nrDEG$log2FoldChange[match(gene$SYMBOL,rownames(nrDEG))]
head(gene)
geneList=gene$logFC
names(geneList)=gene$ENTREZID
#按照logFC的值来排序geneList
geneList=sort(geneList,decreasing = T)
head(geneList)
以上即完成了数据的准备工作,开始进行GSEA分析
#以KEGG Pathway示例
library(clusterProfiler)
#clusterProfiler包的妙用
kk_gse <- gseKEGG(geneList = geneList,
organism = 'hsa',
nPerm = 1000,
minGSSize = 10,
pvalueCutoff = 0.9,
verbose = FALSE)
#提取GSEA-KEGG结果
tmp=kk_gse@result
kk=DOSE::setReadable(kk_gse, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
tmp=kk@result
pro='pair-pre-post/pair-pre-post_gsea'
write.csv(kk@result,paste0(pro,'_kegg.gsea.csv'))
#按照enrichment score从高到低排序,便于查看富集通路
kk_gse=kk
sortkk<-kk_gse[order(kk_gse$enrichmentScore, decreasing = T),]
head(sortkk)
dim(sortkk)
#write.table(sortkk,"gsea_output2.txt",sep = "\\t",quote = F,col.names = T,row.names = F)
#可以根据自己想要的通路画出需要的图
library(enrichplot)
gseaplot(kk_gse, "hsa04510")
gseaplot2(kk_gse, "hsa04510", color = "firebrick",
rel_heights=c(1, .2, .6))#改变更多参数,为了美观
#同时展示多个pathways的结果。
#画出排名前四的通路
gseaplot2(kk_gse, row.names(sortkk)[1:4])
#上图用的是ES排名前4个画图,也可以用你自己感兴趣的通路画图
# 自己在刚才保存的txt文件里挑选就行。
paths <- c("hsa04510", "hsa04512", "hsa04974")
paths <- row.names(sortkk)[5:8]
paths
gseaplot2(kk_gse, paths)
#这里的GSEA分析其实由三个图构成,GSEA分析的runningES折线图
# 还有下面基因的位置图,还有所谓的蝴蝶图。如果不想同时展示,还可以通过subplots改变。
gseaplot2(kk_gse, paths, subplots=1)#只要第一个图
gseaplot2(kk_gse, paths, subplots=1:2)#只要第一和第二个图
gseaplot2(kk_gse, paths, subplots=c(1,3))#只要第一和第三个图
#如果想把p值标上去,也是可以的。
gseaplot2(kk_gse, paths, color = colorspace::rainbow_hcl(4),
pvalue_table = TRUE)
#最后的总结代码
gseaplot2(kk_gse,#数据
row.names(sortkk)[1:5],#画哪一列的信号通路
title = "Prion disease",#标题
base_size = 15,#字体大小
color = "green",#线条的颜色
pvalue_table = TRUE,#加不加p值
ES_geom="line")#是用线,还是用d点
当然,这里不知道具体需要什么通路,就全部都画出来
# 这里找不到显著下调的通路,可以选择调整阈值,或者其它。
down_kegg<-kk_gse[kk_gse$pvalue<0.1 & kk_gse$enrichmentScore < -0.4,];down_kegg$group=-1
up_kegg<-kk_gse[kk_gse$pvalue<0.1 & kk_gse$enrichmentScore > 0.4,];up_kegg$group=1
dat=rbind(up_kegg,down_kegg)
colnames(dat)
dat$pvalue = -log10(dat$pvalue)
dat$pvalue=dat$pvalue*dat$group
dat=dat[order(dat$pvalue,decreasing = F),]
library(ggplot2)
library(enrichplot)
gesa_res=kk@result
###画出每张kegg通路
lapply(1:nrow(down_kegg), function(i){
gseaplot2(kk,down_kegg$ID[i],
title=down_kegg$Description[i],pvalue_table = T)
ggsave(paste0(pro,'_down_kegg_',
gsub('/','-',down_kegg$Description[i])
,'.pdf'))
})
lapply(1:nrow(up_kegg), function(i){
gseaplot2(kk,up_kegg$ID[i],
title=up_kegg$Description[i],pvalue_table = T)
ggsave(paste0(pro,'_up_kegg_',
gsub('/','-',up_kegg$Description[i]),
'.pdf'))
})
GSEA-GO
GO和KEGG分析流程基本相同,除了函数名和变量名的变化
ego <- gseGO(geneList = geneList,
OrgDb = org.Hs.eg.db,
ont = "ALL",
nPerm = 1000, ## 排列数
minGSSize = 100,
maxGSSize = 500,
pvalueCutoff = 0.9,
verbose = FALSE) ## 不输出结果
go=DOSE::setReadable(ego, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
go=ego@result
pro='test_gsea'
go_gse=go
sortgo<-go_gse[order(go_gse$enrichmentScore, decreasing = T),]
head(sortgo)
dim(sortgo)
write.table(sortkk,"gsea_output2.txt",sep = "\\t",quote = F,col.names = T,row.names = F)
#可以根据自己想要的通路画出需要的图
library(enrichplot)
gseaplot2(go_gse,#数据
row.names(sortgo)[1:5],#画那一列的信号通路
title = "Prion disease",#标题
base_size = 15,#字体大小
color = "green",#线条的颜色
pvalue_table = TRUE,#加不加p值
ES_geom="line")#是用线,还是用d点
write.csv(go,file = 'gse_go.csv')
其他可视化方法
气泡图
dotplot(kk,split=".sign")+facet_wrap(~.sign,scales="free")

条形图
down_kegg<-kk_gse[kk_gse$pvalue<0.1 & kk_gse$enrichmentScore < -0.4,];down_kegg$group=-1
up_kegg<-kk_gse[kk_gse$pvalue<0.1 & kk_gse$enrichmentScore > 0.4,];up_kegg$group=1
dat=rbind(up_kegg,down_kegg)
colnames(dat)
dat$pvalue = -log10(dat$pvalue)
dat$pvalue=dat$pvalue*dat$group
dat=dat[order(dat$pvalue,decreasing = F),]
library(ggplot2)
g_kegg<- ggplot(dat, aes(x=reorder(Description,order(pvalue, decreasing = F)), y=pvalue, fill=group)) +
geom_bar(stat="identity") +
scale_fill_gradient(low="blue",high="red",guide = FALSE) +
scale_x_discrete(name ="Pathway names") +
scale_y_continuous(name ="log10P-value") +
coord_flip() + theme_bw(base_size = 15)+
theme(plot.title = element_text(hjust = 0.5), axis.text.y = element_text(size = 15))+
ggtitle("Pathway Enrichment")
g_kegg

网络图
library(ggplot2)
library(enrichplot)
cnetplot(kk,showCategory= 5, foldChange= geneList, colorEdge="TRUE")
#colorEdge不同的term展示不同的颜色,如果希望标记节点的子集,可以使用node_label参数,它支持4种可能的选择(即“category”、“gene”、“all”和“none”).
ego2<-pairwise_termsim(ego)
emapplot(ego2, showCategory= 10, color="pvalue", cex_category=1, layout="kk")
#cex_category调节节点大小,showCategory展示条目个数
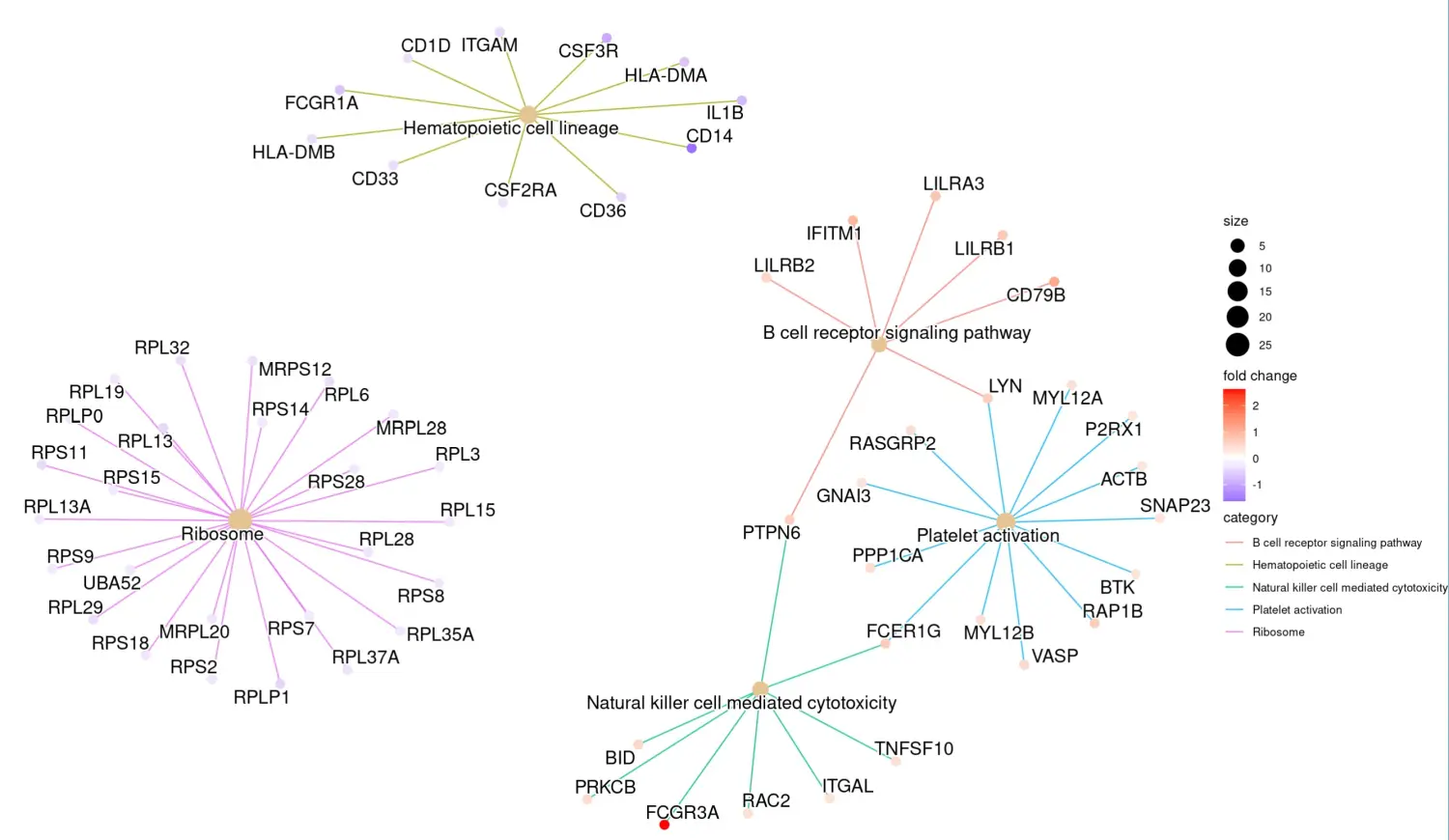
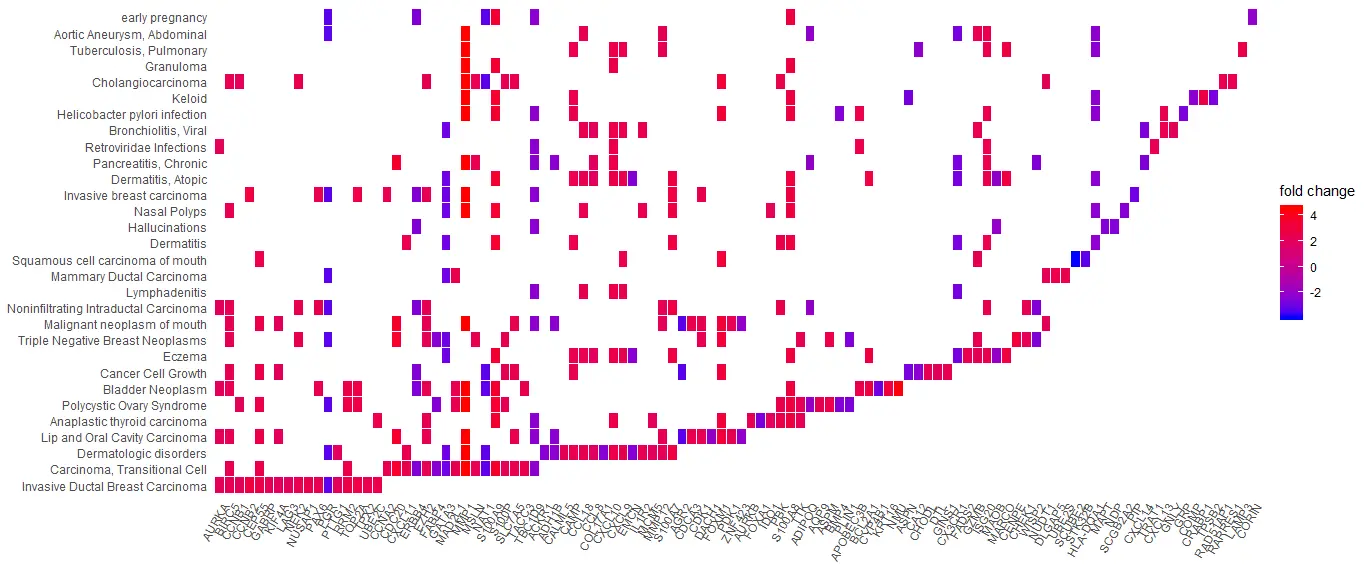
Heatmap-like functional classification
热图与cnetplot类似,但将关系显示为热图。如果用户希望显示大量的重要术语,则基因概念网络可能会变得过于复杂。热图可以简化结果,更容易识别表达模式。
UpSet Plot
upsetplot是cnetplot的一种替代方法,用于可视化基因和基因集之间的复杂关联。它强调不同基因之间的重叠。
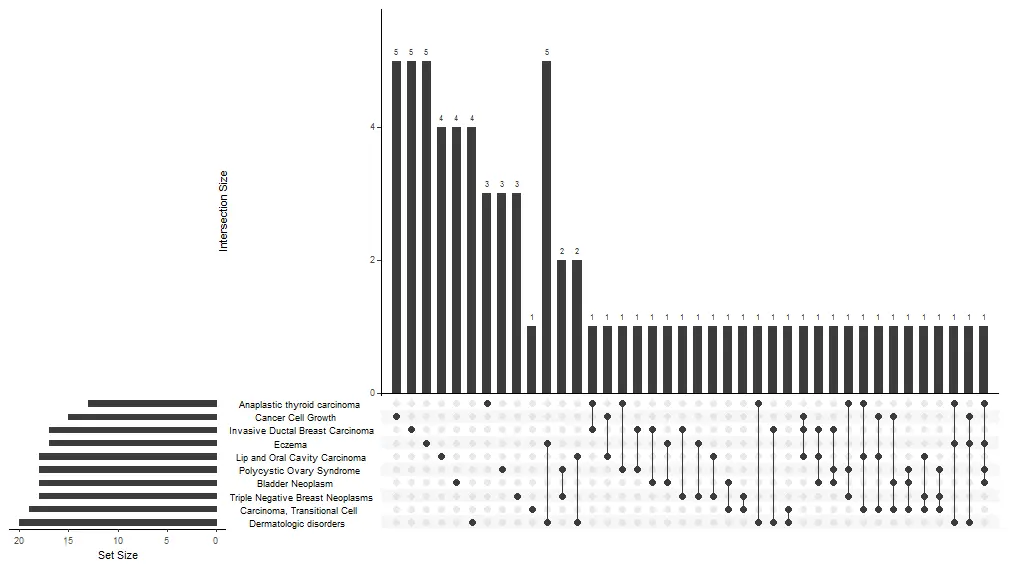
对于过表达分析,upsetplot将计算不同基因集之间的重叠,如图上所示。对于GSEA结果,它将绘制不同类别的褶皱变化分布(例如,路径的独特性,不同路径之间的重叠)。
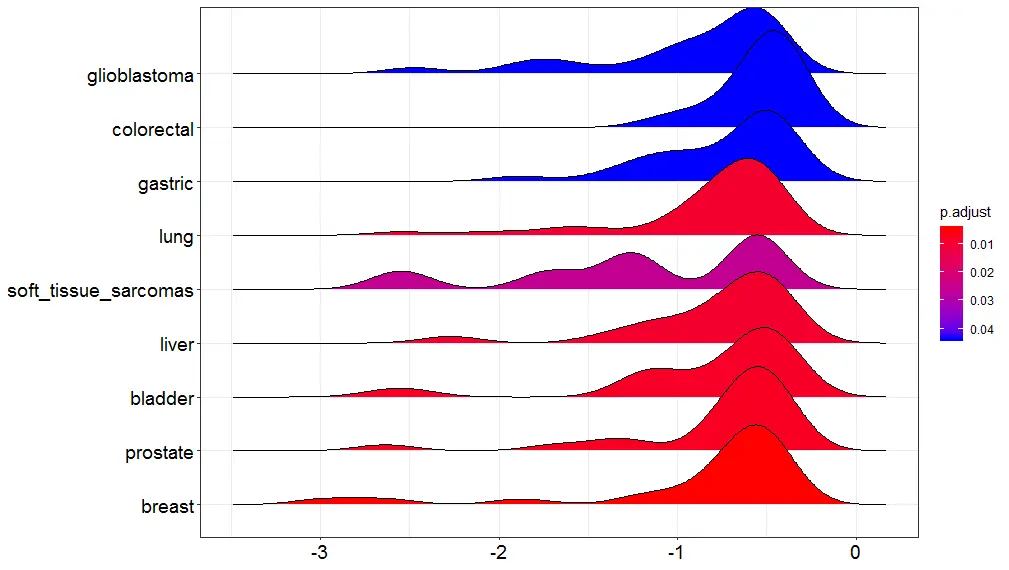
ridgeline plot for expression distribution of GSEA result
ridgeplot将可视化GSEA富集类别的核心富集基因的表达分布。它帮助用户解释向上/向下调节的路径。
ridgeplot(edo2)
参考资料:
