【5.1.1】Kallisto原理及应用
一、Kallisto简介
首先,这款软件是2016年发表在NBT上的一款RNA-seq的计数软件,文章标题为《Near-optimal probabilistic RNA-seq quantification》(http://dx.doi.org/10.1038/nbt.3519) 这款软件对比TopHat+cufflinks,Hisat+HTseq等流程组合,就时间上要快很多。 而该软件的核心思想是省略了将原始数据fastq文件比对到擦靠基因组上,然后再计数这一过程,取而代之的是直接将fq文件的reads比对参考转录组上并且直接计数
1.1 Kallisto算法
该算法认为,将fq文件的reads比对到参考基因组的具体位置,然后根据位置信息进行计数数没有实质性意义的,而该算法采用的是伪比对的方式:即直接将fq文件的reads比对参考转录组上并且直接计数
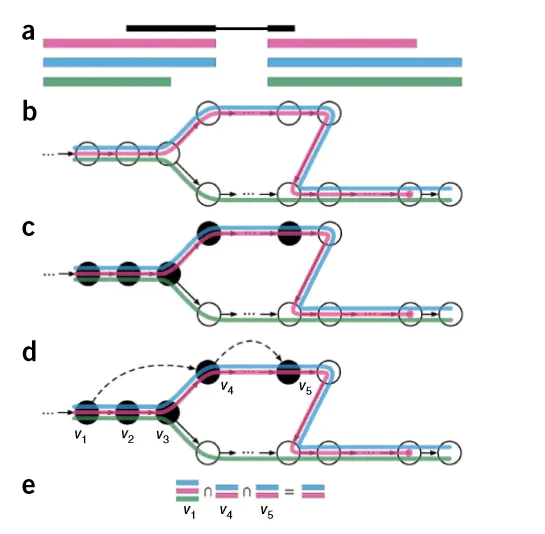
首先,作者先要根据参考转录组建立索引,即可哈希的关系。 然后,作者利用k-mer基于德布鲁因图的方法将fq文件的reads比对到参考转录本上,并且确定哪些k-mer属于哪一个转录本
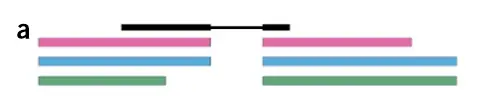
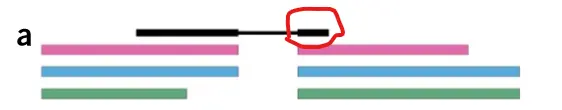
如上图所示,黑色部分表示fq文件的reads,而彩色部分表示具有overlap的转录本的外显子部分 然后利用k-mer根据德布鲁因图将fq文件的reads比对到这些转录本上去
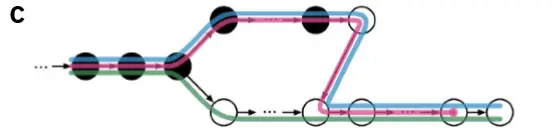
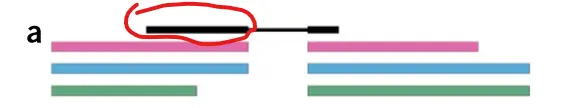
那么上图黑色部分(fq文件reads的前半部分)主要比对到了红色和蓝色这两个转录本上面,图C fq文件reads的前半部分
而白色部分(fq文件reads的后半部分)主要比对到了红色,蓝色和绿色这三个转录本上面,图C fq文件reads的后半部分
那么基于上述结果,我们取一个交集:
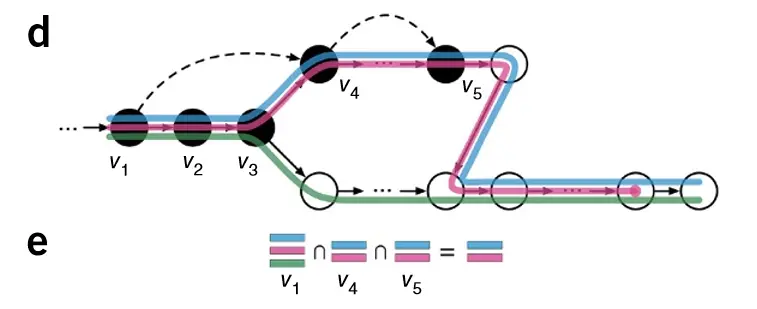
那么,该条reads就计数为红,蓝这两个转录本上了,该软件就利用这个原理进行计数,就省去了比对到参考基因组上这一过程
二、Kallisto具体应用
首先建立参考转录本的索引:
kallisto index -i refMrna.idx refMrna.fa
计数
#单端
kallisto quant -i transcripts.idx -o output -b 100 --single -l 180 -s 20 fastq
#双端
kallisto quant -i refMrna.idx -o output -t 8 -b 100 fastq_1 fastq_2
三、我的案例
git clone https://github.com/pachterlab/kallisto
cd kallisto
mkdir build
cd build
cmake ..
make
make install
cd /data4/aNEO_data/mice/Mus_musculus/UCSC/mm10/Sequence/Chromosomes/
kallisto index -i ref_mm10.idx ref_mm10.fasta
kallisto quant -i index -t 120 -o output pairA_1.fastq pairA_2.fastq
参考资料
- https://www.jianshu.com/p/0e5dfdf56034
- https://www.jianshu.com/p/5828d1d060aa
- 《Near-optimal probabilistic RNA-seq quantification》(http://dx.doi.org/10.1038/nbt.3519)
- https://pachterlab.github.io/kallisto/manual
药企,独角兽,苏州。团队长期招人,感兴趣的都可以发邮件聊聊:tiehan@sina.cn
![]() 个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
