cutadapt
我们对测序数据进行QC,就三个大的方向:
- Quality trimming
- Adapter removal
- Contaminant filtering
当我们是双端测序数据的时候,去除接头时,也会丢掉太短的reads,就容易导致左右两端测序文件reads数量不平衡,有一个比较好的软件能解决这个问题,我比较喜欢的是cutadapt软件的PE模式来去除接头!尤其是做基因组或者转录组de novo 组装的时候,尤其要去掉接头,
一、安装
pip3 install cutadapt
或者:
sudo python3 -m pip install cutadapt
其他安装,可以参考:
https://cutadapt.readthedocs.io/en/stable/installation.html#system-wide-installation-root-required
二、用法
参考资料: http://cutadapt.readthedocs.io/en/stable/guide.html
基本用法举例:
# 去掉3’端的接头
# -a 3’接头
# -o 输出,o是output的意思
# -j 选择几个核
cutadapt -j 10 -a AACCGGTT -o output.fastq input.fastq
# 也可以处理gz压缩格式的文件
# 支持 gzip (.gz), bzip2 (.bz2) 和 xz (.xz).
cutadapt -a AACCGGTT -o output.fastq.gz input.fastq.gz
# 去除5’端的接头
# -g 5'接头
cutadapt -g ADAPTER -o output.fastq.gz input.fastq.gz
# 去除poly-A尾,如去除100个及以上个A
# instead of writing ten A in a row (AAAAAAAAAA), write A{10}
cutadapt -a "A{100}" -o output.fastq input.fastq
# quality triming
# The -q (or --quality-cutoff) parameter can be used to trim low-quality ends from reads before adapter removal.
# By default, only the 3’ end of each read is quality-trimmed.
cutadapt -q 10 -o output.fastq input.fastq
# 去除多个 3’接头
cutadapt -a TGAGACACGCA -a AGGCACACAGGG -o output.fastq input.fastq

Keep in mind that Cutadapt removes the adapter that it finds and also the sequence following it, so even if the actual adapter sequence that is used in a protocol is longer than that (and possibly contains a variable index), it is sufficient to specify a prefix of the sequence(s).
cutadapt的结果
开头为:
Sequence: 'ACGTACGTACGTTAGCTAGC'; Length: 20; Trimmed: 2402 times.
中间的信息为:
No. of allowed errors:
0-7 bp: 0; 8-15 bp: 1; 16-20 bp: 2
The adapter, as was shown above, has a length of 20 characters. We are using a custom error rate of 0.12. What this implies is shown above: Matches up to a length of 7 bp are allowed to have no errors. Matches of lengths 8-15 bp are allowd to have 1 error and matches of length 16 or more can have 2 errors. See alsothe section about error-tolerant matching.
结尾的部分:
Finally, a table is output that gives more detailed information about the lengths of the removed sequences. The following is only an excerpt; some rows are left out:
Overview of removed sequences
length count expect max.err error counts
3 140 156.2 0 140
4 57 39.1 0 57
5 50 9.8 0 50
6 35 2.4 0 35
7 13 0.3 0 1 12
8 31 0.1 1 0 31
...
100 397 0.0 3 358 36 3
更多关于结果的解读,可以查看cutadapt User Guide: how-to-read-the-report
三、案例
3.1 探查adapter
首先用fastqc软件对测序数据进行简单检测,看看有什么接头:

既然fastqc能探测到你的接头,说明它里面内置了所有的接头序列,在github可以查到:
https://github.com/csf-ngs/fastqc/blob/master/Contaminants/contaminant_list.txt
或者:Download common Illumina adapters from https://github.com/vsbuffalo/scythe/blob/master/illumina_adapters.fa
TruSeq Universal Adapter AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT Illumina Small RNA 3p Adapter 1 ATCTCGTATGCCGTCTTCTGCTTG
最严重的一般是TruSeq Universal Adapter , 而且它检测到你其它的接头可能就是这个 TruSeq Universal Adapter 的一部分而已~!
我们可以先用cutadapt去除试一下 cutadapt软件支持对PE 测序数据的处理,基本的用法是:
cutadapt -a ADAPTER_FWD -A ADAPTER_REV -o out.1.fastq -p out.2.fastq reads.1.fastq reads.2.fastq
-a和-A是左右端测序数据的3端接头,-g和-G是左右端测序数据的5端接头。
支持fastq和fasta格式的gz压缩文件,必要时用-f参数指定测序文件数据格式即可。
cutadapt软件支持对PE 测序数据的处理,基本的用法是:
cutadapt -a ADAPTER_FWD -A ADAPTER_REV -o out.1.fastq -p out.2.fastq reads.1.fastq reads.2.fastq
-a和-A是左右端测序数据的3端接头,-g和-G是左右端测序数据的5端接头。 支持fastq和fasta格式的gz压缩文件,必要时用-f参数指定测序文件数据格式即可。
我自己的实际例子如下:
python ~/miniconda2/pkgs/cutadapt-1.10-py27_0/bin/cutadapt
-a AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT -a ATCTCGTATGCCGTCTTCTGCTTG
-A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT -A CAAGCAGAAGACGGCATACGAGAT
-e 0.1 -O 5 -m 50 -n 2 --pair-filter=both
-o read_PE1.fq -p read_PE2.fq test1.fastq test2.fastq >& log.txt
参数解释如下:
- 两个-a 参数后面接的是两种接头,两个-A参加后面接的是同样的两个接头的反向互补序列!
- 加上 -n 2 是因为有两个接头,我的测序数据里面有可能一个read里面含有两种接头,所以需要检测两次。
- -e 0.1 -O 5 -m 50 是标准参数,自己看readme就好了,其中-m 设置为50 是表示去除接头后如果read长度小于50我就不要了,因为我是PE150测序的,这也就是为什么要用PE模式来去除接头,保证过滤后的reads还是数量继续平衡的。
去除了接头之后的文件再用fastqc软件跑一下,很明显可以看到 接头去除成功啦!
四、原理
4.1 什么是接头
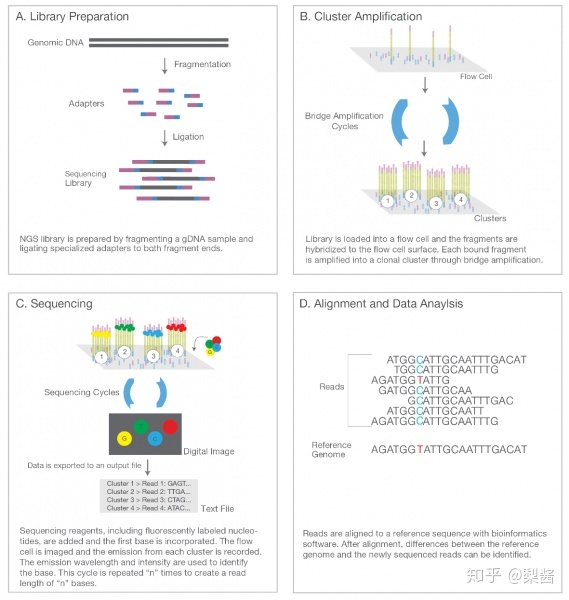
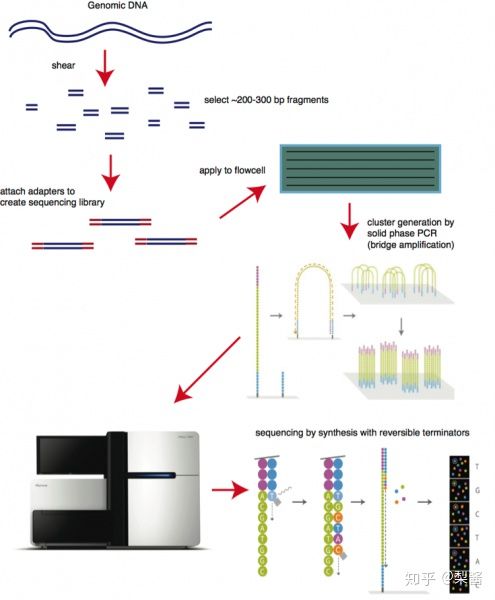
4步获取测序数据
在了解接头之前,简要介绍一下获取测序数据的步骤,如上图主要分为4步:
- 制备文库
- PCR扩增
- 测序
- 比对分析
去除接头adapter,则是第4步“比对分析”中的一个小环节。
那什么是接头呢?
在第1步“制备文库”时,我们会在样品DNA片段末端加上adapter(接头)。
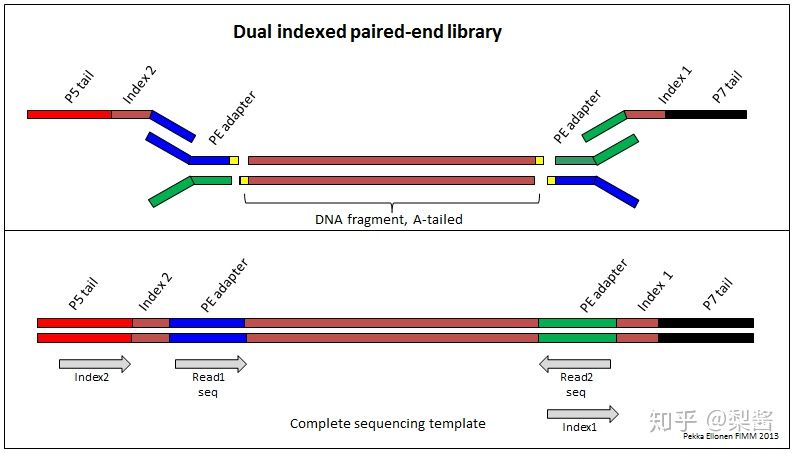
接头方式1,图片来自FIMM,侵删
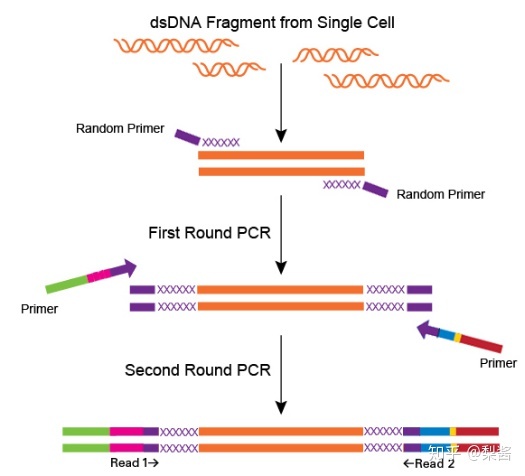
加接头方式2,图片来自贝瑞基因,侵删
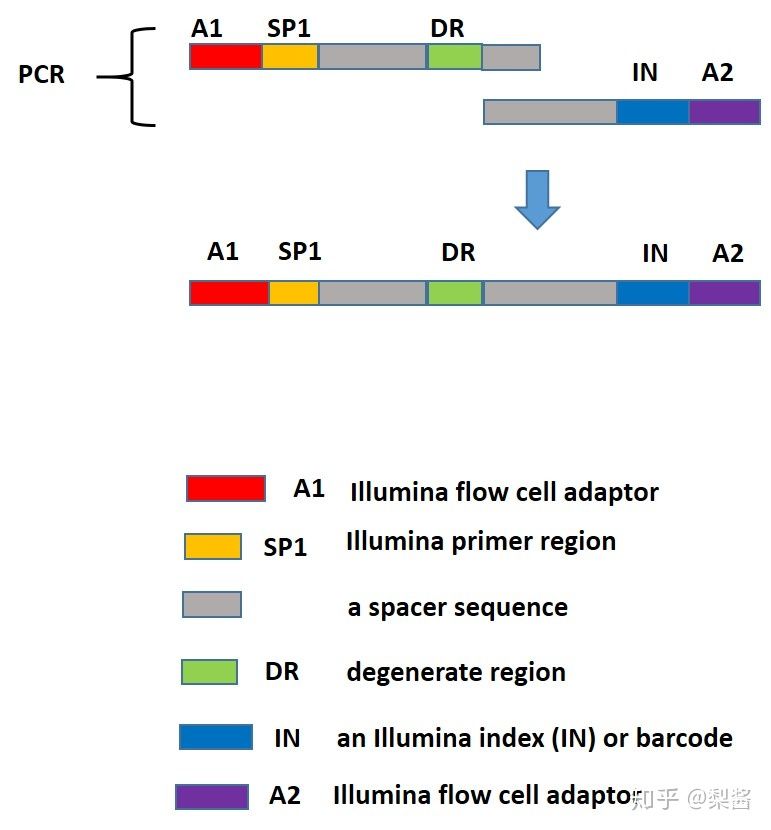
加接头方式,图片来自网站blackberryaurora,侵删
注意!加接头的方式有多种。
There are a number of different ways to prepare samples, all preparation methods add adapter to the end of DNA fragment. illumina 一般,一个DNA片段两端会有两个FlowCell的adapter,还有一个index。
这些接头在第2步“PCR扩增”时,会和固定在flowcell的接头互补配对。样品DNA片段便被吸附在flowcell表面,从而进行桥式PCR扩增成DNA簇。
怎么知道数据来自哪个样品?
科学家们的方法是,给DNA片段加上标签(barcode/index)。就像超市里货架上用来区分不同商品的条形码。
在Solexa多重测序(Multiplexed Sequencing)过程中会使用Index来区分样品,并在常规测序完成后,针对Index部分额外进行7个循环的测序,通过Index的识别,可以在1条Lane中区分12种不同的样品。
为什么要扩增成DNA簇?
因为这样才能产生亮度达到CCD可以分辨的荧光点。
注:
- flowcell上有8条lane(泳道),每条lane可以直接物理区分测序样品。
- 1次run(单次上机测序反应)最多可以同时上样8条Lane,大概产生4G-75G测序通量。
- 每条Lane中排有2列tile,合计120个小区。每个小区上分布数目繁多的簇结合位点。
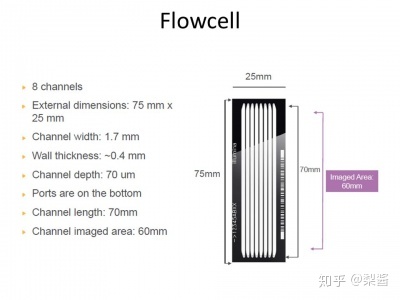 Flowcell. (图片来自illumina,仅供学习,侵删)
Flowcell. (图片来自illumina,仅供学习,侵删)

FlowCell for HiSeq 2000 (图片来自hackteria.org)
最后为了得到所需数据,就得去掉这些人为在DNA片段两端加上去的接头和标签。
补充说明下单端测序和双端测序的区别:
What is the difference between “Single-End” and “Paired-End” reads? Single-End Read: When the sequencing process only occurs in 1 direction (utilizing Read Primer 1). Paired-End Read: If two separate read cycles occur in both directions (utilizing both Read Primer 1 and 2). This kind of read will provide data about both sides of the fragment of interest (Blue). If the fragment size is consistent you will also be able to predict that both the forward and reverse reads will be a known distance from each other. This data can be used to help the software map the reads more accurately.
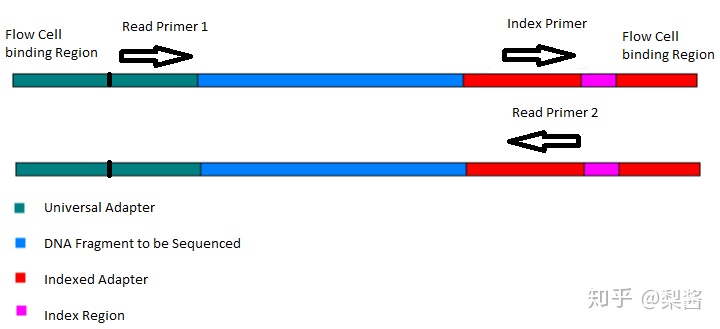
图片来自Frequently Asked Questions. TUCF Genomics
五、我的案例
参考资料
