【4.3.8】预测蛋白质抗原决定簇的半经验方法(Kolaskar and Tongaonkar antigenicity)
预测网页:http://tools.iedb.org/bcell/
Description: A semi-empirical method which makes use of physicochemical properties of amino acid residues and their frequencies of occurrence in experimentally known segmental epitopes was developed to predict antigenic determinants on proteins. Application of this method to a large number of proteins has shown by the authors that the method can predict antigenic determinants with about 75% accuracy which is better than most of the known methods.
Scale:
A C D E F G H I K L M N P Q R S T V W Y
1.064 1.412 0.866 0.851 1.091 0.874 1.105 1.152 0.93 1.25 0.826 0.776 1.064 1.015 0.873 1.012 0.909 1.383 0.893 1.161
对蛋白质上由实验确定的抗原性位点进行的数据分析表明,如果疏水性残基Cys,Leu和Val存在于蛋白质表面,则它们更有可能是抗原性位点的一部分。 开发了一种半经验方法,利用氨基酸残基的理化特性及其在已知的已知片段表位中的出现频率来预测蛋白质上的抗原决定簇。 该方法对大量蛋白质的应用表明,我们的方法可以预测抗原决定簇,其准确度约为75%,比大多数已知方法要好。 该方法基于单个参数,因此使用非常简单
一、前言
B细胞表位在蛋白质抗原上的描述近年来引起了几位科学家的关注[1-5]。鉴定蛋白质上的表位对于诊断目的以及肽疫苗的开发将是有用的[6]。为了帮助实验人员,霍普和伍兹开发了一种预测抗原决定簇的方法[7]。 Hopp和Woods的方法已被修改,以考虑到抗原性位点在蛋白质表面且大多数表面残基具有抗原性这一事实。最近,Parker等人使用了三个参数-亲水性,可及性和柔韧性 -通过合成图预测B细胞表位。与Hopp和Woods的方法相比,该方法改进了对抗原决定簇的预测,但是错过了一些实验观察到的决定簇。另一方面,Welling等人已经根据表位中氨基酸的存在频率计算了每种氨基酸的抗原性值,并使用这些值来预测表位[9]。这些工作者使用的数据库非常小,仅包含来自20种蛋白质的606个氨基酸。因此,计算出的频率值具有较大的误差。我们已经使用实验性抗原决定簇数据和氨基酸的理化特性得出了一个参数。使用该单一参数,已开发出一种预测抗原决定簇的方法,其准确度约为75%。
二、材料和方法
2.1 抗原性的计算(Ap值, antigenic propensity)
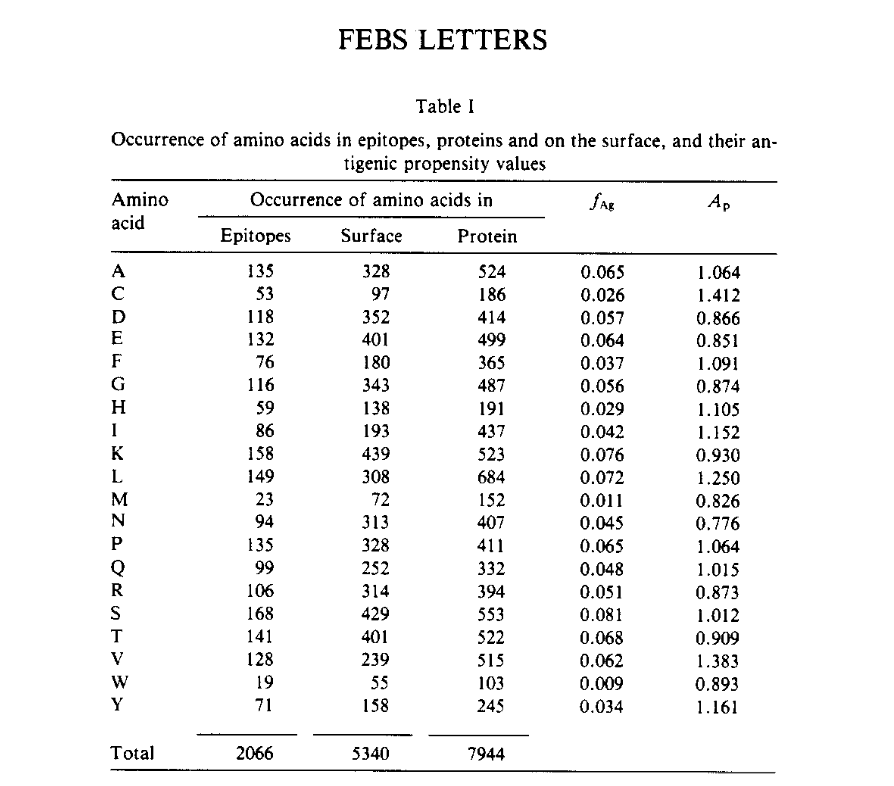
通过实验确定了34种不同蛋白质中的169个抗原决定簇(蛋白质清单请参见表II)。在这169种已知抗原决定簇中,有156个决定簇的氨基酸残基少于20个。这项研究156个实验测定的抗原决定簇含有2066个氨基酸残基。
- 利用这些数据,可以计算出抗原决定簇中每种氨基酸的出现频率(fag, frequency of antigenic determinants )。
- 可以通过以下步骤预测蛋白质表面的残基:亲水性(Ph),可及性(Pa)和柔韧性(Pf),使用Parker等人的表II中给出的20个蛋白质氨基酸的值。在使用这些参数值的给定蛋白质中,计算得出的平均,Ph,Pa和Pf,从N端到C端的每一个重叠的七肽。这些值分配到每个片段的中间(i + 3)残基,如果 Pai > pa,或 Phi > ph或 Pfi >pf,则认为该残基在表面,其中pa,ph,pf是蛋白质的平均值。使用此数据,可以计算出氨基酸在表面(fs)上的出现频率。
- 使用以下关系式计算每种氨基酸的抗原倾向(Ap)值:
Ap = fag/fs
2.2 预测抗原决定簇的算法
-
计算从蛋白质的N末端到C末端的重叠七肽平均抗原倾向值Ap 。 将这些平均值分配给段中的第四个(i + 3)残基。(7个残基求平均值,将值赋予第4个残基)
-
确定蛋白质的平均抗原倾向 Ap_mean 值。
-
如果 Ap_mean ≥ 1.0,则那些具有 Ap ≥ 1.0的残基称为潜在的抗原残基。 如果 Ap_mean <1,那么那些具有Ap ??? 的残基被称为潜在的抗原残基。(这里的没看懂!!!!)
-
挑选抗原决定簇的条件是六个连续的残基必须满足步骤3的条件。 该算法用于预测34种蛋白质的抗原决定簇,对此可获得一些实验结果。 该计算机程序可用于PC兼容系统。
三、结果与讨论
从表I可以看出,Ser,Lys,Thr,Glu和Ala在抗原决定簇中的发生频率较高。但是,这些频率值可能会引起误解,因为已知其中有几个氨基酸也以较高的频率出现在总蛋白中(这可以从表I中看出,该表给出了所考虑的34种蛋白中的氨基酸)。我们知道当今很少蛋白质的完整抗原结构是已知的,因此fAg可以改变。由于这种变化,此处给出的A值是暂定值。然而,有趣的是,对于疏水性氨基酸Cys,Val和Leu,Ap值非常大。因此,每当这些残基出现在表面上时,它们很可能是抗原决定簇的一部分。从目前的分析来看,这是非常重要的发现。表II中给出了将该算法应用于34种蛋白质的结果。我们的方法比Parker等人的方法遗漏的站点数量要少。 (请参阅表II)。从表II中可以看出,在我们的169种实验已知的抗癌决定簇中,我们的方法正确地提取了122种抗原决定簇,平均准确度约为75%。因此,我们的方法可以准确地选择抗原决定簇。通过我们的方法预测的其他位点数量在经过深入研究的蛋白质中很少,如IL-3,外壳蛋白/ TMV,促红细胞生成素和肌红蛋白,其中缺失的位点分别为0、2、2和1。根据这些结果,我们认为通过我们的方法预测的大多数其他位点可能是致癌因素。因此,简而言之,开发了一种基于单参数的半经验方法,该方法明智地利用了氨基酸残基的理化性质和实验数据,可以预测抗原决定簇,并且其准确性已通过应用于大量蛋白质而得到检验。

参考资料
- Kolaskar and Tongaonkar antigenicity scale Reference: Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990 Dec 10;276(1-2):172-4.
