【4.5.9】CDR聚类--north
抗体的互补决定区(CDR)的先前分析已集中在每个环的少量构象构象上。这主要是Chothia及其同事最近一次于1997年的工作结果。由于抗体的广泛应用,我们重新审视了六个CDR环构象的聚类,并提供了目前可用的大量结构信息。在这项工作中,我们通过消除低分辨率结构和具有高B因子或高构象能量的CDR来谨慎使用高质量的数据集。我们使用了基于方向统计(directional statistics)的距离函数和具有亲和力传播(affinity propagation)的有效聚类算法。利用300多种非冗余抗体结构的数据集,我们能够涵盖L1,L2,L3,H1和H2的28种CDRS长度组合(例如,我们的CDR长度命名法中的L1长度11或L1-11)。 Chothia分析仅涵盖20个CDR长度。其中只有四个具有不止一个构象簇:
- 其中两个可以容易地通过基因来源(小鼠/人类;k/λ)来区分,
- 而一个可以简单地通过Pro残基的存在和位置来轻易地区别(L3-9 )。
因此,使用Chothia分析不需要通常假定的复杂的“结构确定残基”集。在我们的28个CDR长度中,有15个具有多个构象簇,其中10个的Chothia分析只有一个规范类。对于非H3 CDR,我们总共有72个群集;可以基于基因来源和/或序列将大约85%的非H3序列分配给构象簇。我们发现,基于H3锚残基Arg / Lys94和Asp101的存在或不存在,关于bulged [构型]与nonbulged [构型]构象的早期预测没有成立,因为所有四个组合均导致大多数构型凸起。因此,增加的数据大大增强了早期的分析。我们相信,新的分类将导致抗体结构预测和设计的改进方法。
一、前言
抗体三维结构的预测是提高其亲和力,稳定性和作为治疗剂的适用性的重要步骤。考虑到重链可变(VH)域和轻链可变(VL)域的框架的保守结构,结构生物信息学中的许多注意力都集中在参与结合抗原的互补决定区(CDR)上。 Chothia,Lesk,Thornton等人在1980年代和1990年代的研究集中于为VH域的六个CDR环[H1,H2和H3,VL结构域的L1,L2和L3]确定少量经典结构(canonical structures)的想法。 。11987年首次提出的中心假设是,免疫球蛋白中的大多数高变区都具有一小部分离散的主链构象,我们称之为canonical structure,并且少数关键残基可用于预测新的CDR序列可能属于哪个构象类别。在进一步的研究中,Al-Lazikani等人,Martin和Thornton,Oliva等人,Wilmot和Thornton,6Shirai等人,和Kuroda等人定义了基于环长度的经典结构,并且在某些情况下,某些环长度的构象不同。在某些位置上的残基特别是甘氨酸,脯氨酸,芳族残基以及氢键供体和受体被认为是造成构象差异的原因。在1997年的研究中,Chothia和他的同事发现了总共25个规范的类,这是由于可用的结构数量更多
Chothia等使用抗体环和序列的手动聚类来定义其规范分类。 Martin和Thornton在1996年使用定量聚类方法(quantitative clustering)进行自动分类方案。他们在内部坐标空间中进行了聚类分析,然后对笛卡尔坐标空间中的结构组进行了聚类后合并[使用均方根偏差(RMSD)]对观察到的CDR进行分类。 在某些情况下,他们观察到,尽管loop在顺序上可能更接近一种Chothia规范类,但它在结构上属于另一种。 他们指出,这是对先前研究的更多基于序列分析的限制。
有许多研究专门针对结构多样的重链H3 CDR中发现的结构基序。将H3高变区域分为环的torso区域和head。他们发现躯干通常具有两种构型之一,即鼓起或扩展(bulged or extended)的β-sheet,以及head的可能构型然后,该区域受躯干残基的结构限制。 Oliva等也根据结构将H3环分成几组。他们使用几何字母定义环构象,如Wilmot和Thornton所述。Shirai等人通过检查确定了一系列的sequence结构关系,然后将它们转换为一组规则以对H3结构进行分类。特别是,他们相信torso区是否存在盐桥,如Morea等人所定义,导致该区域的凸起或扩展构象。4Kuroda等。后来修改了他们的H3 sequence-structure规则列表,并提供了更多的H3结构
对于非H3环,其构象的最新综合分析是在1996-1998中进行的。随着可用抗体结构数量的大量增加,我们决定重新进行抗体CDR构象的分析,以查看基于17种结构或少于60种结构的规范分类是否成立,以及是否可以确定新的分类。在本文中,我们根据当前的蛋白质数据库(PDB)更新所有六个CDR区域的分类。我们过滤掉了低分辨率的结构,具有高B因子或高构象能量的loop以及冗余序列。总共使用337条独特的重链和311条独特的轻链来构建抗体环的结构数据库。与Chothia的分析不同,我们发现将CDR分为CDR类型(L1,L2等)和loop长度最为直观。我们简称为CDRS长度组合简称为CDR-lengths。例如,CDR L1的loop长度为11,我们将其指定为L1-11。然后,我们使用具有二面角距离函数的亲和力传播聚类方法,将聚类应用于特定CDR-length组合的所有环路的构象。 我们发现,Chothia等人发现的大多数规范构象。现在可用的300+抗体结构中的许多都发生了这种情况。我们已经鉴定出总共72个构象簇,其中大多数在两个或多个抗体结构中观察到。我们将根据较小的数据集将我们的结果与以前的抗体loop分类进行详细比较。
二、结果
2.1 数据集
如资料和方法中所述,我们使用手动策划的多序列比对来构建VH和VL域的隐藏Markov模型(HMMs)。 我们使用这些模型搜索整个PDB序列集,以识别具有抗体可变域的所有PDB链。 共有923个抗体PDB条目,其中包含至少一个定义了所有主链原子位置的高变环。 由于许多PDB条目的不对称单元包含同一抗体的一个以上拷贝,并且由于其他PDB条目包含一个以上的抗体(抗独特型,anti-idiotypes),因此这些文件中包含具有VH结构域的1232链,具有VL结构域的1304链 ,以及30条在一条单链中同时具有VH域和VL域的链(scFv片段)。 排除低分辨率结构(>2.8 Å)和NMR结构后,剩余703个条目包含882个VH域,953个VL域和26条scFv链。
我们对CDR的定义不同于最常用的Kabat和Chothia方案。 我们选择了这样的定义:使用Honegger和Plckthun获得的结构比对,使每个环的锚点(anchors)(紧靠环之前或之后的残基)相对于框架包含紧密聚集的构象。我们还选择了这样的位置: 末端残基和C末端残基在结构上彼此相对,无论它们出现在相邻的β-链(CDR2和CDR3)还是不同的β-折叠(CDR1)中。 在可能的情况下,我们还选择使用VL和VH链中同源位置的定义。

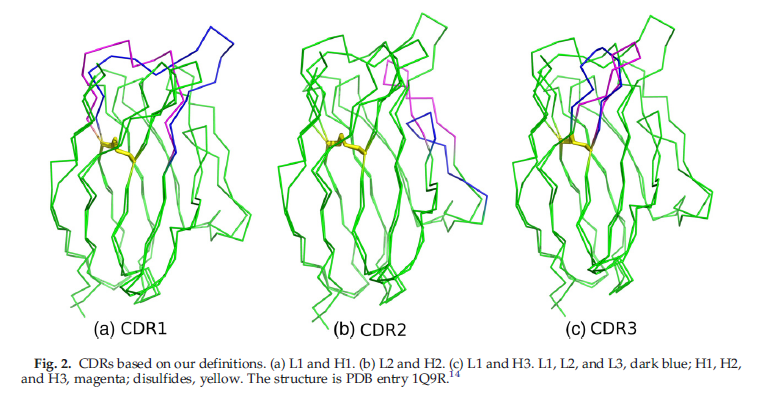
CDR起始和终止位置周围的序列基序如图1所示。我们从紧接链内二硫键保守半胱氨酸的位置开始,并将它们定义为H1,L1,H3和L3的N末端残基。对于CDR1和CDR3环,我们基于Cα位置选择C-末端,在VL和VH域之间的差异最小,这在结构上也与N-末端的深度大致相同。在这四种情况下,C末端是保守的芳香族残基,这些残基是每个结构域疏水核心的一部分。我们选择L2起始位点作为Chothia使用的相同位点,因为它出现在我们已经选择的CDR1位点的对面,并位于β-2链的末端,并以此将H2起始位点置于相同位置。我们将H2的末端紧紧放在H2的N末端的短β-2链中。但是,VL中的该区域并不总是β-链,并且对于多个其他残基具有序列和结构多样性。我们选择的L2 C末端与Martin-Thornton定义相符。该L2定义在其C末端比H2定义多包含三个位置。 VH和VL结构域(来自PDB条目1JU12)与所示CDR的叠加如图2.14所示。请注意,每个环的N末端和C末端位于VH结构域和VL结构域之间的同源位置, L2除外(图2b)。
我们应用了许多标准来筛选出不确定或不确定构象的loop。这些包括缺少坐标的环,具有高B因子的骨架原子,具有不是脯氨酸的顺式残基的残基[包括PDB条目1OCW13(分辨率,2.0 -E)和10个非Pro顺式残基,包括4个Pro顺式残基在H1]中的结构,以及那些具有高骨架构象能的结构(由我们最近发表的Ramachandran概率分布确定)。由于某些抗体的结构已被多次确定,因此其余结构在序列上是高度冗余的。通过用六个CDR的序列表示每个可变域结构,我们为每个序列选择了最高分辨率的结构。我们还删除了少数具有相对于所有其他结构异常的构象的环,这些环定义为与数据集中的所有其他结构至少具有一个主干二面角90°。表1中显示了应用了这些滤波器中的每一个之后数据集中每个CDR的loop数。表2中给出了所得数据集中每个CDR的不同循环长度的计数。


2.2 CDR环构象的亲和力聚类
我们针对CDR,环长度和cis-Strans配置的每种组合分别运行了亲和力聚类算法(affinity clustering algorithm)。作为聚类的示例,我们在图3中显示了L1-12聚类的Ramachandran分布。此CDR长度包括12个具有唯一序列的结构,聚集成大小5、5和2的三个构象。我们划分了Ramachandran图进入如图4所示的标记区域,以便通过构象标记簇。在这个定义中,B是β-折叠区域,P是聚脯氨酸II,A是α-螺旋,D是δ-区域(α-螺旋附近,但Φ的负值更大),L是左手螺旋,G是γ-区域(Φ >0°,不包括L和B区域)。根据这些定义,群集1(蓝色点)的中位环具有BPABPBPAADBB构型,群集2(洋红色点)的中位环具有BPABPPPLLPBB构型,群集3(绿色点)的中位环具有BPPAADAAPPBB构型。簇1与簇2的区别主要在于残基8、9和10,分别具有构象AAD和LLP。
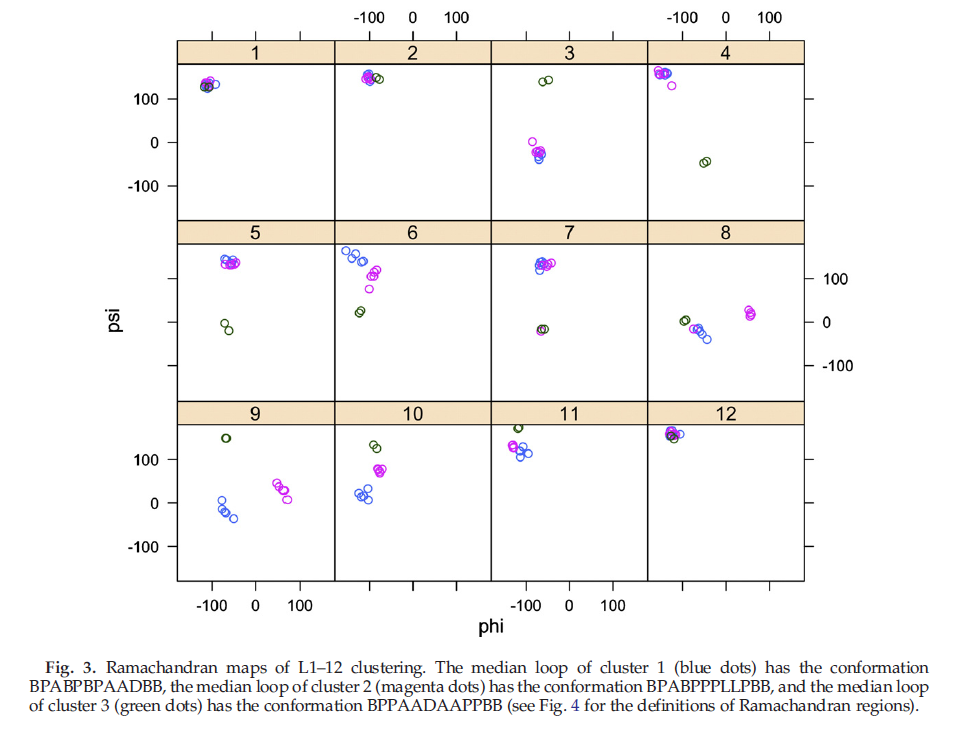

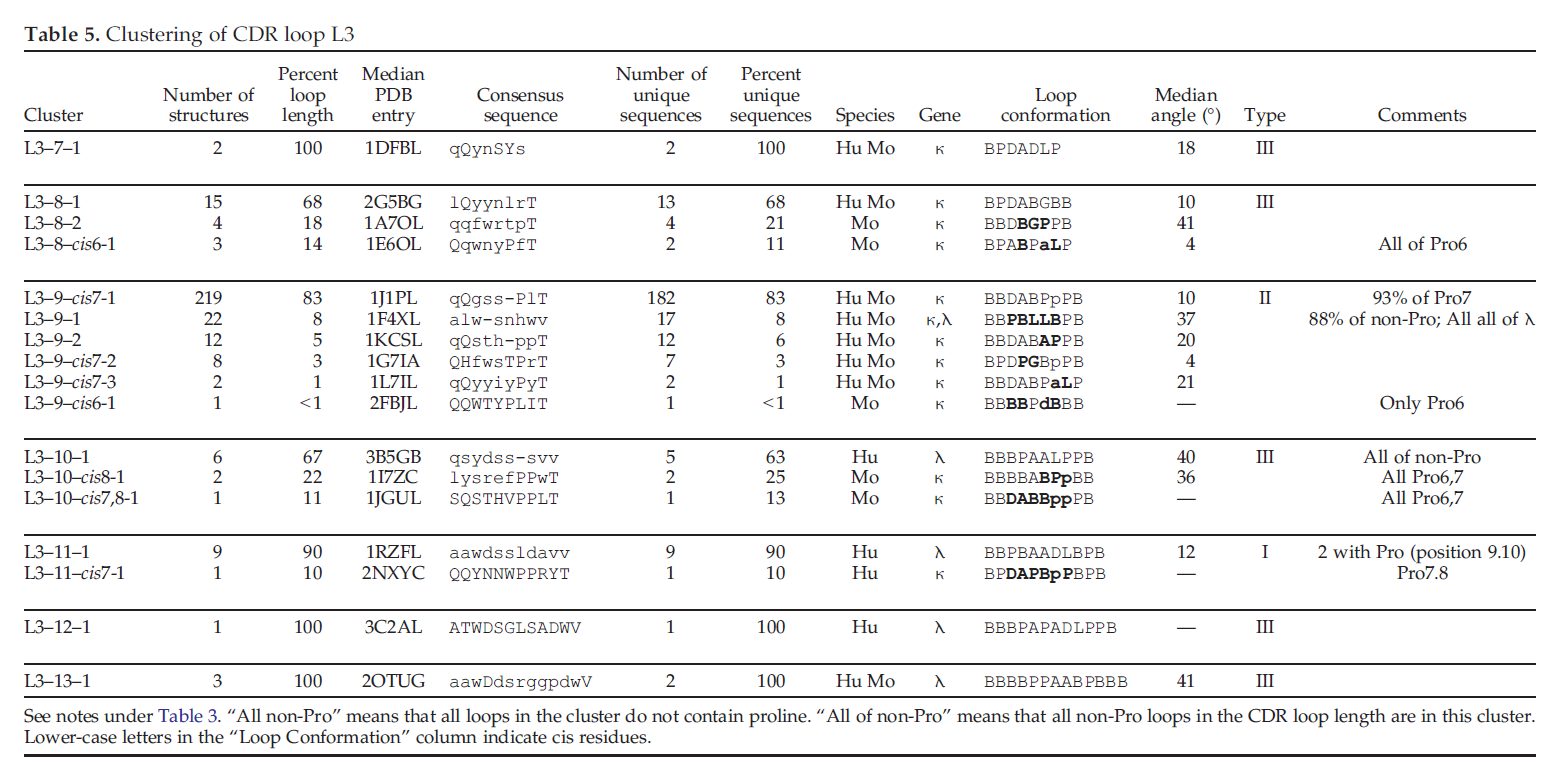
表3、4、5、6和7分别显示了CDR L1,L2,L3,H1和H2的聚类结果。 表3中显示了H3 loop的躯干区域的聚类。在每个表中,给出了每个循环长度的结果; 对于每个簇,给出了结构数和百分比,唯一序列数,中位环结构的PDB条目,共有序列以及就Ramachandran构象而言的中位环构象。




在讨论每个CDR的聚类结果之前,我们可以观察到抗体环长度的三种不同类别或类型,这些类型或类型指示从其序列可预测CDR长度构象的程度:
-
I型:对于第一种类型,某些CDR-length组合的环具有一个构象,该构象形成所有结构或至少大部分结构。当输入亲和力算法时,结果是一个构象簇或一个大簇和少量外围构象。大型群集必须分布相当紧凑。因此,该CDR长度具有可预测的结构。如果至少有10个独特序列,并且在最大构象簇中有超过85%的结构,我们认为CDR长度就是这种类型。
-
类型II:可预测的CDR长度–第二种CDR-length组合具有多种可能的结构,但是每个簇紧密分组,并且在序列上与其他簇明显不同。我们在这种类型中包括一些环,这些环的构象簇很容易通过某些框架残基的同一性来预测,即使不同簇中的环没有明显不同的序列也是如此。要属于这种类型,循环必须在每个较大的簇中至少具有四个唯一的序列,两个或多个簇,并且其成员关系可以通过loop的序列(或通过某些框架的标识)预测到的85%以上残留物)。
-
III型:不可预测的CDR长度– 对于某些CDR长度,结构预测可能很困难或统计上不确定。出于多种原因可能会发生这种情况。首先,亲和力传播过程可能会将大多数结构放入少量高度分散的簇中或放入大量非常小的簇中。第二,可能没有太多的结构对聚类有足够的信心。在某些情况下,可能会建议一个确定簇的序列基序,但是数据不足以令人信服。对于其他CDR长度,结构可以很好地聚类为离散的构象,但它们的序列几乎没有系统变异。对于这些CDR长度,未知结构环的结构预测可能取决于与其他CDR,抗原或框架的相互作用。
我们依次讨论每个CDR。
L1
L1根据我们的定义,L1的环长度可为10至17个残基。 L1环的大多数长度为11或16,分别具有57和50个唯一序列。 L1的聚类分析结果显示在表3中。
L1的几种长度为I型,这意味着单个构象非常重要。 CDR长度L1-10是其中之一,具有22个总结构中的20个(所有小鼠k)属于单个构象。两者的中位构象是BBABPBABBB与BBABPBPGPB,其主要区别在于残基位置7和8,涉及这些残基之间肽键的翻转。这是两个同源结构之间的常见且相对较小的差异。 L1-16也属于I型,所有68个结构都属于一个群集。 L1-17也是单簇CDR长度,所有21个结构都具有相似的构象。这些环与它们的中值结构的平均距离为每个二面角10°(请参见表3)。这些小的值表示紧密的聚类。

L1-11属于II型,具有三个可供选择的构象,可通过CDR序列或某些构架残基的同一性轻松预测。我们将这些群集称为L1-11-1,L1-11-2和L1-11-3。我们首先查看了从每个簇中的唯一序列得出的序列logo,以确定序列是否可以区分这些簇;这些如图5所示。簇L1-11-3在5和6位氨基酸分布非常不同,簇L1-11-1和L1-11-2具有[SDNE] [IV],而L1-11-3具有[ILA] [GPS]。 L1-11-3序列全部来自人Vλ; L1-11-1和L1-11-2具有非常相似的氨基酸分布,它们来自人和小鼠的Vk链。正如Al-Lazikani等人所指出的,仅基于四个结构,L1-11-1和L1-11-2之间的结构差异是由于位置71的构架不同(Chothia编号; CDR L3开始之前的18个残基; Honegger-P1uckthun编号系统10中的残基89)。当第71位的残基为Phe时,67个此类结构中的63个(94%)位于簇L1-11-1中。 Thr位置71的所有8个结构以及Gly位置71的所有结构均在L1-11-1中。在Tyr位于71位的50个结构中,其中48个(96%)位于簇L1-11-2中。簇L1-11-1中的环从CDR残基7的羧基氧到残基68(L3之前的21个残基)的酰胺氢原子形成氢键。在属于簇L1-11-2的环中,CDR的残基7和残基8之间的酰胺键的方向相反。这将残基8的酰胺氢原子引向71位酪氨酸残基的羟基氧原子,形成氢键。这些交互如图6所示。

其余的L1长度只有少量可用结构和序列,包括L1-12(12个结构,12个序列,3个簇),L1-13(11个结构,11个序列,2个簇),L1-14(18个结构,12个序列,2个簇) )和L1-15(13个结构,11个序列,2个簇)。即使在这里,仍有一些残基可以区分这些簇。但是,由于观察到的数量少,我们不能确信这些功能将始终具有预测性。因此,我们将它们归类为III类。例如,群集L1-12-3(鼠标Vλ;)的序列与L1-12-1(鼠标Vk)和L1-12-2(人和小鼠Vk)的序列非常不同。 5个L1S-12-1成员中有4个具有Tyr71,而所有5个L1-12-2成员均具有Phe71。 L1-13的两个簇(均为人Vλ)通过位置2和5的序列很容易区分,L1-13-1的前五个残基的序列基序为[ST] G [ST] [SAT] [ST],而L1-13-2具有序列基序TRSSG。 L1-13-2第5位的Gly可能对此残基具有γ构象( Φ,Ψ= + 70°,+ 160°)。 L1-14的两个簇具有完全不同的序列。 L1-14-1簇中的人类序列具有共有序列RSStGavTtsNYAN(完全保守的残基为大写),而L1-14-2的小鼠序列具有共有序列TgtssnvgGynyVs。 L1-14-1第5位的Gly可能偏爱该残基的-3构象。最后,群集L1-15-2仅具有两个鼠标Vk成员,它们彼此仅相差一个残基。 L1-15-1和L1-15-2的构象在位置7-9不同,分别具有序列[DE] [YSFN] [YFD]和STS。
L2
L2的聚类分析结果如表4所示。已知结构的L2 loop只有两种长度:L2-8和L2-12。 L2-8有308个结构; 其中,由159个独特序列组成的290个(占94%)属于具有BLLDPPPP构象的多数簇。 第二个最常见的簇具有9个结构,其中位结构具有BLLDPPPA构象,仅在最后一个残基处与主要构象不同。 还有另外三个非常小的群集。 我们认为L2-8是I型,即实际上仅具有一种构象。
L2-12在两个簇中仅包含四个结构,每个簇只有一个唯一的序列。 第一个是人类前B细胞受体的结构,而第二个是小鼠Vλ结构体。 由于序列很少,因此该loop属于III型。
L3
L3的聚类分析结果如表5所示。L3环的长度为7到13,其中85%L3环的长度为9。L3-9的最大簇,占该CDR长度的83%。一种在第7位含有顺式脯氨酸的氨基酸,我们将其命名为L3-9-cis7-1。还有两个带有cis7的非常小的群集,两个群集是全反式的,一个群集具有cis6。仅通过脯氨酸残基的位置(如果有的话)就可以很好地预测L3-9环的结构。如果预测所有带有Pro7的L3-9环都在簇L3-9-cis7-1中,则此预测是235次的正确219次或93.2%的时间(对于唯一序列为93.8%)。其余的10个位于其他cis7群集中,6个位于全反式群集L3-9-2中。如果L3-9完全不存在Pro,则25个中的22个(或88%)位于L3-9-1群集中。因此,L3-9是II型,通常在结构上是可预测的。四个最大簇的每个代表结构的叠加参见图7。

L3的三个额外CDR长度包含一个以上的簇,并且所有三个长度均为III类(即数量很少):L3-8,L3-10和L3-11。 Pro在位置6的所有三个L3-8 loop都属于L3-8-cis6-1簇。有两个全反式簇,它们没有彼此区别的序列特征。对于L3-10,所有没有脯氨酸的环都属于全反式簇L3-10-1。两个簇L3-10-cis8-1和L3-10-cis7,8-1都在位置7和8上包含两个脯氨酸。单个L3-11-cis7-1结构在位置7和8上具有Pro,而全反式L3-11-1结构都没有。
三个环长度(L3-7,L3-12和L3-13)只有一个构型和一个或两个唯一序列,因此属于III型。后两个CDR长度为λ序列。
H1
H1的聚类分析结果显示在表6中。CDRH1的长度为12至16,长度也为10。最短和最长的H1序列来自骆驼科抗体。 CDR长度的H1-13代表92%的H1环,并由单一构象控制。 簇H1-13-1包含306个结构中的267个(占87%),构象为PPBLBPAAABPBB,最小归一化中位角为13°(参见表5)。 因此,它是类型I。剩余的39个结构分布在11个不同的集群中,具有广泛的可能结构。 它们之间不存在明显的序列差异,只是其中的三个仅针对骆驼科抗体出现。 H1的其他CDR长度都存在于单个簇中。 但是,它们每个都包含少于10个唯一序列。 因此,这些CDR长度为III型。 铁汉 15:45:37
H2
H2的聚类分析结果如表7所示。对于H2,存在两种共有的环长(H2-9和H2-10),每个都有多个聚类,还有三个环长,每个环只有一个聚类(H2-8,H2-12,和H2-15)。对于H2-9,81个结构中的77个(或95%)属于H2-9-1簇,最小归一化中位角为10°(请参见表7)。因此,它是I型。所有H2-9人类序列均在H2-9-1中。簇H2-9-1和H2-9-3都在位置6处具有L构象,而簇H2-9-2具有D构象。与此一致的是,H2-9-1和H2-9-3在此位置上主要有Gly(H2- 9-1中有一些Asp),而H2-9-2具有Phe和Val。
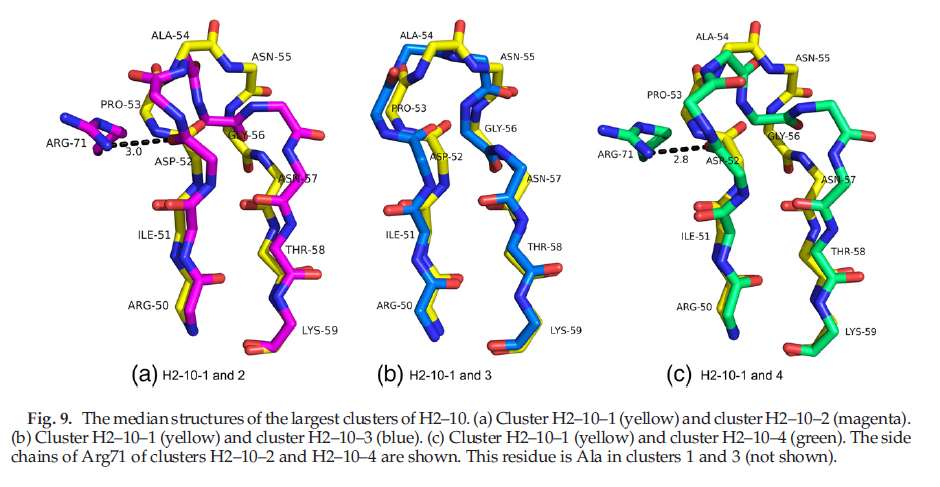
CDR长度的H2-10代表所有H2环的67%。它分为两个大类(占结构的68%和19%)和七个小得多的类。我们检查了前四个簇的序列徽标,发现在loop的中间几个位置,Gly和Pro的位置有不同的模式,如图8所示。左旋的L或G构象没有一个位置是完全可预测的,因此我们根据每个聚类中的唯一序列使用HER20创建了隐藏的Markov模型,然后将每个loop分配给了得分最高的集群。对于具有BBPAADLPBB构象的簇H2-10-1,可以正确预测155个结构中的130个(或84%)。对于具有BBPAALABBB构象的H2-10-2簇,正确预测了42个簇中有30个(或71%)结构存在于簇中。 H2-10-3和H2-10-4的预测不是很好,但人口要少得多。具有BBBPGALPBB构象的H2-10-3具有正确预测的11个结构中的6个。最后,具有BBPPLLABBB构象的H2-10-4仅能正确预测7个结构中的2个。但是,总的来说,H2-10的环序列相对于其簇的HMMs的得分擅长预测序列的簇成员。

对于H2,Tramontano等人注意到构架残基对确定环构象的影响,特别是残基71的身份(Chothia编号,H3开始前的25个残基; Honegger和PlUckthun10编号82)。他们使用我们的CDR定义分析了H2-9,H2-10和H2-12(它们的长度分别为3、4和6);但是,在1990年,它们分别只有2、3和2个结构。我们决定对此进行调查,以了解它是否拥有更大的数据集。对于H2-9,无论位置71(Val和Arg),他们仅发现一种构象。我们还发现只有一个构象有效(H2-9- 1 = 77 of 81结构)。位置71不能帮助区分H2-9-2和H2-9-3(数据未显示)与H2-9-1。对于H2-10,Tramontano等人发现两个构象:两个具有Arg71的结构类似于我们的簇H2-10-2,一个具有Ala的结构类似于我们的簇H2-10-1。在表9中,我们显示了H2-10的列联表,列中第71位的残基不同,行中的H2-10簇不同。对于H2-10,我们总共有227个结构和196个独特序列。我们也有9个构象聚类,而不是2个,尽管只有前两个聚类很高。如果我们仅从位置71预测结构所属的群集,则将在表9的每一列中分配具有最高编号的群集。例如,如果位置71是Ala,则可以预测群集H2-10-1,我们将得到67个正确的作业和13个错误的作业。如果位置71是Arg,我们将预测簇H2-10-2并获得58个正确分配中的38个。如果我们在每个群集中添加最大数量,则可以正确预测186个LOOP(或80%),与上面讨论的隐藏markov模型相当(群集1-4中78%的lop)。如表所示,主要的小疏水残基(A,I,L或V),小的极性残基(S和T)或Q,在这种情况下,环主要属于簇H2-10-1(161次中的143次,或90%);如果残基为R或D,则该残基属于簇H2-10-2(59次中的39次,即66%)。簇2、3和4与簇H2-10-1的中位结构的叠加示于图9。簇H2-10-2和簇H2-10-4在71位具有Arg,并且与CDR残基3的羰基氧键合。

最后,对于H2-12,所有26个结构都属于单个紧密簇,最小归一化中位角为8°,因此将此loop定为I型,而两个非常小的CDR长度H2-8和H2-15也只有一个簇(类型III)。
H3
H3的已知环结构的长度非常不同,从5到26个残基不等,大多数(86%)在7到16个残基之间。较短的环可以很好地聚集在一起,但是比例较少(表2)。较长的环形成一些具有较高自相似值的大型群集,但是这些群集与中位数的距离非常大。一些群集在Ramachandran地图的不同区域中(例如A和L区域)有残基。在自相似度较低的情况下,簇的数量变得非常大,并且簇的大小变得非常小。因此,它们不太可能具有预测价值。
由于这些困难,许多分析已将H3拆分为对应于其N端和C端的torso或锚定区域,并在循环的转折处将其拆分为head或end区域,将骨架区域分为两组:bulged和 nonbulged,或kinked和 extended。 我们对包含H3的前三个残基的七个残基进行了亲和力传播聚类(红色代表图1中的N-末端区域)和H3的最后四个残基(图1中的C-末端区域为红色,在左侧另加一个)。这些七个残基的不连续肽的聚类结果显示在表8中。对于H3躯干聚类,总共有八个聚类。 H3-anchor-1群集覆盖了大约三分之二的结构,而前四个群集覆盖了约95%。前三个聚类显示在图10中。单个残基位置的列联表未显示H3躯干聚类的可预测性(数据未显示)远远超过第一个聚类中的65%。
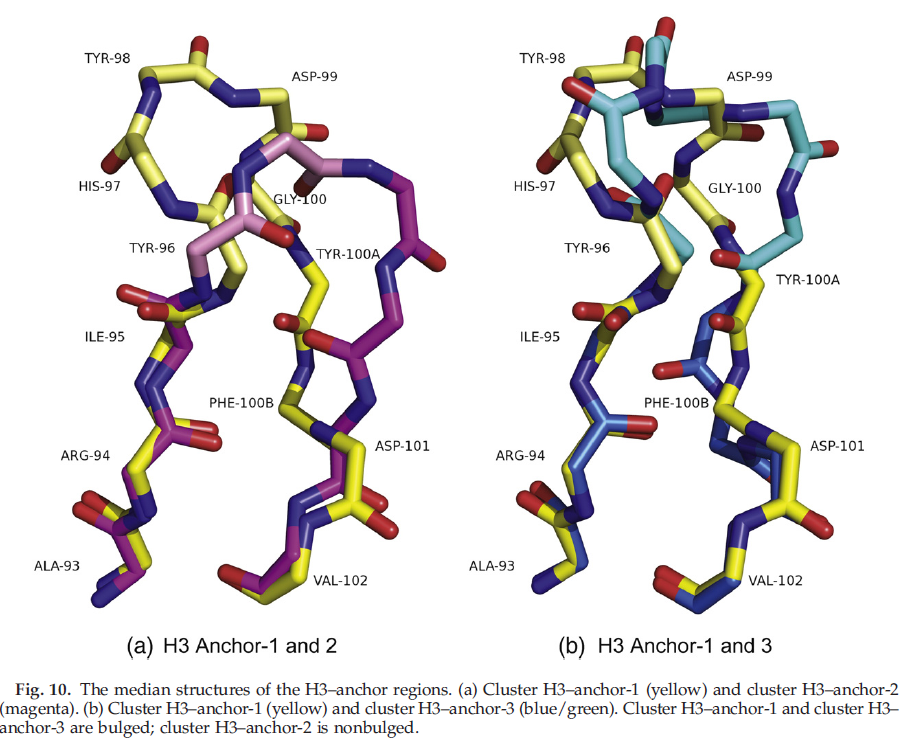
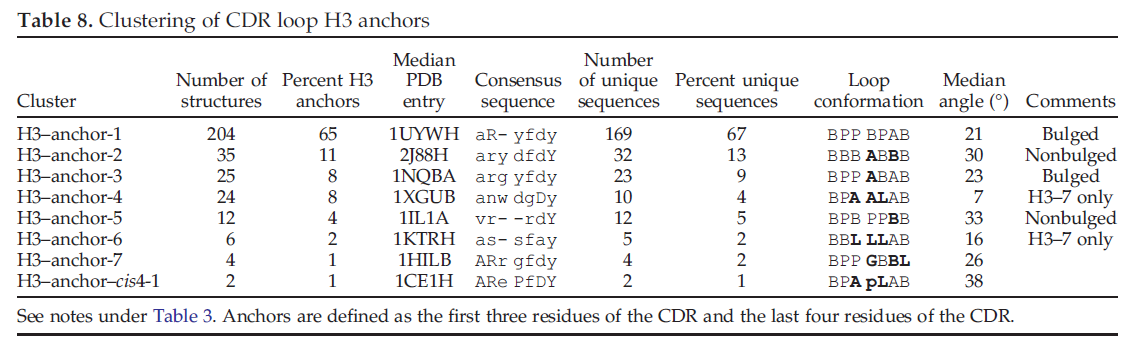
我们针对不同长度的H3 loop检查了这些簇的分布。结果显示在表10中。我们将H3-7 loop包括在H3-锚点聚类中,即使不能期望它们与较长环的躯干区域很好地聚类。确实,这些loop主要分为三个簇,彼此独立:H3-anchor-4,H-Sanchor-6和带有cis4的簇。将少量H3-7结构放置在群集H3-anchor-1中。有趣的是,对于其他长度,分布在某种程度上取决于长度。对于H3-8(仅5个结构),其中2个,1个和2个结构分别位于H-anchor-1,H3-锚-2和H3-anchor-5中。 H3-9(26个结构)是唯一的非凸起H-Sanchor-2簇的H3 CDR长度。对于长度为10到14的H3,74-79%的结构属于H3-anchor-1。但是,对于长度15和16,则92%的结构属于H3-anchor-1,其余部分位于H3-anchor-5群集中。对于长度超过16的H3循环,71%属于H3-anchor-1,其余部分属于H-Sanchor-3。这些频率在从17到26的整个loop长度上是一致的(数据未显示)。

2.3 与Chothia和Martin-Thornton聚类的比较
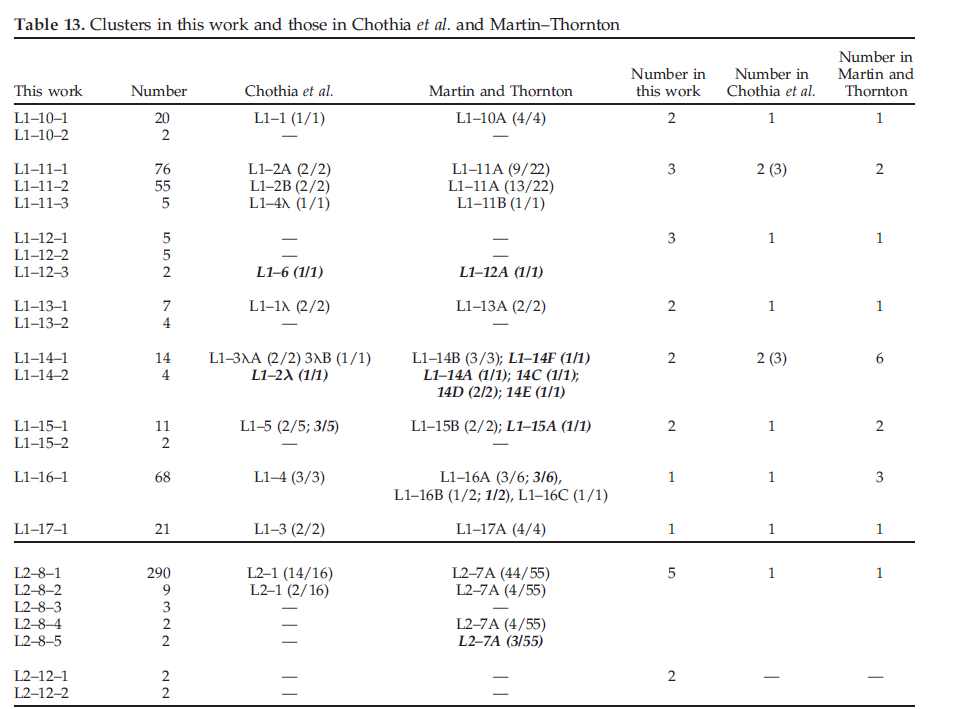
以前有一些关于抗体环结构分类的研究。本研究的聚类结果概括了Al-Lazikani等人和martin和Thornton所发现的许多经典构象。我们的构象聚类方法和最新的结构数据库与Al-Lazikani等人,还有Martin和Thornton的结果产生了一些显着差异。我们的聚类与Al-Lazikani等的聚类之间的对应关系。表11、12和13中给出了Martin和Thornton。




我们使用1997年的Al-Lazikani等人的研究来定义Chothia典型构象,因为这是他们以前对抗体的分析中最新,最全面的CDR结构。Chothia等规定,每个CDR的指定规范类都由整数(1、2、3等)组成,与loop的长度无关,并且没有特定的顺序。不同的名称可能是不同长度的环或相同长度但结构不同的环。对λ轻链的CDR进行了分析,并与k链分开编号;在Martin和Thornton之后,我们将它们称为1的λ、 2λ等。某些类被细分为子类,通常是由于一个结构与另一个结构之间的环中两个氨基酸段的翻转。他们将它们指定为A,B等,然后将它们附加到Chothia类名称(例如L1-2A和L1-2B)。对于每个规范类,他们提供了一个或多个适合该类的PDB条目以及这些环的CDR序列及其Φ,Ψ值。对于某些loop,他们仅提供了抗体的名称,我们从这些名称中找到了相应的PDB条目。他们基于总共17个高分辨率结构进行聚类,是通过人工和视觉方式而非计算方式进行的。
Martin和Thornton在二面角空间中进行聚类(使用正弦和余弦矢量),类似于此处执行的聚类,然后基于坐标RMSD合并聚类。他们通过CDR,长度和字母指定它们的聚类。每个不同的构型(即L1-11A,L1-11B,L1-12A等)。他们为每个群集的代表提供了PDB条目,并提供了其群集对57个PDB条目的分配表。
我们的CDR定义与Chothia等人还有Martin和Thornton的定义有些不同。将这些定义应用于样本k,λ和重链序列的比较如图11所示。对于Chothia,我们使用Al-Lazikani等人在研究中给出的示例序列。它们是Kabat定义的CDR中观察到的构象变化的区域,通常在每个末端都带有一个额外的氨基酸,以进行良好的测量。 Al-Lazikani等人描述的区域,并不总是与CDR的“ Chothia定义”相吻合。

如图11所示,Chothia等人(2002年)定义了他们的λ和k CDR1彼此不同。它们的k定义在我们L1定义的N端和C端都短了两个氨基酸。他们的λ定义仅在每个末端短一个氨基酸。它们的L2定义比C端短3个残基,而L3定义比N端短3个残基。与L1 k类似,我们的H1定义在两端比Chothia定义长两个残基,就像我们的H2定义一样。 Martin和Thornton使用了与L1,L3和H2相同的CDR定义。它们的L2在我们的残基之后开始一个残基,而它们的H1在我们的残基之后开始三个残基(我们的序列开始于我们的L1在Cys之后立即发生,而它们的开始在Cys-Xxx-Xxx-Xxx之后开始)。
对于Chothia和Martin-Thornton,我们都使用了他们研究中给出的PDB条目,以将他们的集群与我们的集群进行匹配。在许多情况下,相同的PDB链存在于我们的过滤数据中,并且我们可以进行一一对应。在某些情况下,由于分辨率低,B因子高,构象能量高或去除了冗余序列,我们排除了一些PDB条目或特定的loop。在这些情况下,我们计算了两项研究均引用的PDB条目中的环与相同CDR和相同长度的簇中位数之间的距离函数D。我们将D标准化为循环中残基数的两倍(以说明-U和-H),然后将Eq(2)取反, 计算Φ,Ψ的平均差(以度为单位)。
这些比较的结果对于Chothia数据在表11中给出,对于Martin-Thornton数据在表12中给出。下表提供了这些研究中提及的每个loop指定的部分或全部PDB条目。如果链与我们的集群名称一起列出,则该loop位于我们的集群数据中,并且存在于该loop集群中。如果在PDB链后的括号中给出一个距离,则这是Φ,Ψ与loop簇中值的平均绝对差。在某些情况下,该距离大于25°,我们以斜体将其列出。这样,这些对应关系就不太确定了,并且可能是PDB中该loop的低分辨率或高B因子的结果。在某些情况下会注意到这一点。
Al-Lazikani等,在1997年的研究中列出了超过20个CDR-length组合的25个规范类(表11);如果我们将它们在一个类中的替代构象视为单独的类,则有32个类。应该注意的是,这32个类别中只有3个基于PDB中的五个以上结构,而32个类别中的15个(将近一半)仅基于一种结构。对于大多数规范类,我们可以通过Chothia等人给出的PDB链对集群进行清晰的一对一分配。例如,它们的L1-2A,L1-2B和L1-4-λ集群是我们的L1-11-1,L1-11-2和L1-11-3集群。如上所述,L1-11-3易于从序列上与L1-11-1和L1-11-2区分开,而L1-11-1和L1-11-2由于VL位置71上的残基而彼此不同。
在三种情况下,Chothia等人给出的PDB链的经典类别落入了我们多个集群(cluster)中。对于最大的群集L2-1,L3-1和H1-1会发生这种情况。在这三种情况下,Chothia等人给出的大多数结构属于我们的一个集群,而少数属于另一个集群。由于在这些情况下我们的环较长,所以结构差异可能发生在Chothia等人分析的区域之外。在四种情况下,给定CDR的多个Chothia规范分类中的结构属于我们的一个簇。对于子类L1-3-λA和B,L3-1λA和B,H2-3A和H2S-C会发生这种情况,我们将它们放在单个群集中。
在某些情况下,Chothia代表未出现在我们的数据集中,并且距离我们的中位数结构相对较远。在这些情况下,对我们集群的分配是不确定的。例如,他们的L1-6是低分辨率(3-E)结构,与我们的L1-12-3集群相距52°。他们的L1-2λ尽管它的序列(TGSSSNIGAGHNVK; PDB条目7FABL)显然符合我们的L1-14-2模式,但在我们的数据中,L1-14-2集群与它的最接近的邻居相距很远(43°)。它们在PDB条目7FAB中的L3-1λ B结构也与我们的L3-9-1集群不太接近(46°)。
有趣的是,在20种Chothia CDR-length组合中,只有4种包含一个以上的规范类别:L1-11(L1-2A,B和L1-4λ); L1-14(L1-2λ和L1-2λA,B); L3-9(L3-1,L3-3和L3-1λ A,B,C);和H2-10(H2-2A,B和H2-3A,B,C)。 铁汉 15:45:49
表12中列出了Martin-Thornton群集。他们的研究列出了L1,L2,L3,H1和H2的49个群集。在五个或更多PDB条目中仅观察到八个群集(15%),而在一个PDB条目中仅观察到其中28个群集(57%)。后者中的22个远离我们簇的主位体结构,这些都在表12中突出显示。指出它们是低分辨率还是高构象能,因此使人们怀疑它们是否应列为单独的簇。这些包括L1-14C,D,E,F,L3S-9E,F,H1-10C,D和H2-10D,E,F。在某些情况下,在我们的分析中,Martin-Thornton群集被划分为多个群集,部分原因是我们的loop有时更长(L2和H2)或由于artin和Thornton使用了RSD步骤。例如,它们的L1S11A在我们的L1S11S1和L1S11S2集群之间平均分配。由于两个结构的主链原子之间的RMSD差异较小,martin和Thornton将这两个结构合并为同一簇。由于loop位置7和8处Φ,Ψ角度的巨大差异,我们的算法将两个簇分开。将它们列为同一规范类的A和B构象。尽管有几个结构是我们的L2-8-2,L2-8-4和L2-8-5集群的成员,或者更接近我们的L2-8-1集群,但Martin-Thornton L2-7A集群中的许多都与我们的L2-8-1相对应。同样,它们的L3-9A簇对应于我们的L3-9-cis7-1,但是它们的簇成员之一是对应于我们的L3-9-2的全反式结构。我们还拆分了它们的H2-9A和H2-10A集群。
我们在Martin-Thornton分析中检查了CDR-length组合,发现实际上只有六个具有可以用我们的数据验证的构象簇:L1-11,L1-14,L3-8,L3-9,L3-11和H2- 10(在我们的定义中) 。在Martin-Thornton分析中,其他几个CDR长度具有多个簇,但依赖于分辨率非常低的结构或具有高构象能的结构。例如,它们的L3-10 loop由四个簇组成,但是所有这些簇都具有低分辨率或高构象能。
最后,我们通过在表13中列出我们的簇以及与Chothia和Martin-Thornton数据重叠的PDB链loop数,以另一种方式检查结果。由于我们删除了单例离群值,因此共有72个簇,每个簇至少有两个成员,除非给定CDR和给定长度(例如H2-15-1)或cisStrans配置只有一个结构。我们的集群中有31个拥有五个或更多成员。
在Chothia分析中,我们的四十一个集群没有相应的规范类。因此,我们所拥有的集群数量是Chothia分析中存在的集群数量的两倍以上。这些都是针对Chothia等人提供的PDB中不存在的CDR长度。这些包括L2-12,L3-12,L3-13,H1-10,H1-12,H1-16,H2-8和H2-15。在少数情况下,通常在结构差异较小时(例如,我们的L3-9-1中的L3-1λA和L3-1λ C都包含),我们的集群包含不止一个Chothia规范类。
Martin-Thornton分析中有32个聚类没有相应的聚类,另外10个聚类与它们的聚类之间只有遥远的关系(表13中的斜体),总共有42个。他们的数据集(大多数与Chothia数据中没有的数据集相同),因为分析是在大约同一时间进行的(1996-1997)。我们的某些集群包含多个Martin-Thornton集群;但是,在几乎所有情况下,这些结构均与我们的中位结构相距甚远,因此通常由于分辨率低或构象能量高而被排除在我们的数据集中之外。
2.4 Comparison of H3 torso analysis to Morea et al H3躯干分析与Morea等人的比较
Morea等人提出了基于Chothia编号94和101位置(分别为HoneggerS Pluckthun编号108和137)的残基类型来预测躯干的凸起和非凸起构象的规则。这些是第2位和第6位表7中的7个残基片段。凸起的构型是我们定义中环的最后两个残基的-AB构型,主要是簇H3-anchor-1。非凸起构象具有-BB构象,主要由簇H-Sanchor-2组成。在Morea等人的分析中,鼓起的躯干(bulged torsos)在位置94处具有赖氨酸或精氨酸,而在位置101处通常(但不总是)存在天冬氨酸。对于我们的数据,我们在表14所示的列联表中总结了存在或不存在Lys / Arg94和Asp101的结构数,以及环的状态为凸起或不凸起的情况。
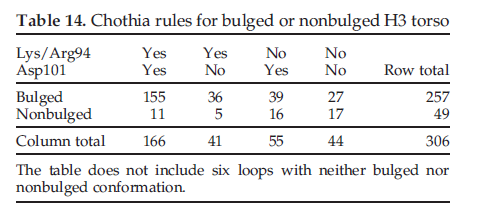
根据Morea等人的观点,如果位置94为Lys / Arg,位置101为Asp,结构凸出。 155个结构具有该序列,并以凸出的构象-AB结尾,而11个结构具有该序列,但不凸出,因此是Morea等人的规则的反例。根据Morea等,如果在残基94上存在Lys / Arg,但在101位上不存在Asp,则该结构仍应凸出。对于具有该序列的36个结构,这是正确的,但对于其余五个结构,则不是这样。如果残基94上不存在Lys / Arg,而101上存在Asp,则认为该结构是非凸出的。但是,我们发现了39个凸起的示例,只有16个非凸起的结构。最后,在他们的研究中,没有观察到同时缺乏94位Lys / Arg和101位Asp的结构。在我们的数据集中,有44个示例,其中有27个是凸起的,有17个不是。六个结构似乎既不适合凸起的构象,也不适合不凸起的构象,因此不予考虑。因此,无论Lys / Arg或94位的其他残基,还是Asp或101位的其他残基,大多数H3躯干结构都凸出。但是,当Lys / Arg位于94位时,有92%的结构凸出。没有Lys / Arg,则有67%的结构凸出。 铁汉 15:45:59
三、讨论
在这项工作中,我们重新探讨了将抗体的六个CDR环的结构聚类的问题。自从Chothia等人以及Martin和Thornton在1996年至1997年的工作以来,尚未完成诸如此类的全面分析。现在,抗体结构的数量至少比以前多5倍(比Chothia使用的抗体多15倍)。因此,对于相同CDR和相同长度的所有其他结构,我们已经能够删除可疑结构(低分辨率或高能主链构象的结构)和异常值。除非它们是给定CDR,长度和cis-trans构型的唯一代表,否则几乎我们所有的簇都由一个以上的结构表示。
当前的分析提出了两个有趣的问题:
- CDR结构可在多大程度上预测序列?
- 是否有较早的分析方法,特别是Chothia canonical类,它们被广泛用作抗体建模和分析的标准构象集?
有了大数据集,我们就开始了开发用于预测CDR环结构的预测方法。对于许多CDR长度,有一个簇代表所有或至少大部分的可用结构。就结构预测而言,在大多数情况下,使用优势簇可能是安全的,除非存在与该簇和或另一个簇相抵触的特定残基。我们试图注释表3-7中的明显差异。在某些情况下,存在明显的结构决定残基(例如L3-9中的Pro)或较大的序列差异(小鼠/人或k/λ序列),这使得鉴定合适的簇相对容易。将CDR构象的统计分析转变为结构预测方法将需要其他工作。但是,我们尝试将可用的CDR-length组合分为不同类型,具体取决于它们是否仅显示一种有效的构象(“ I型”),两种或更多种可以根据CDR序列和/或某些序列在很大程度上预测的构象框架位置(type II),或基于数据不足的一种或多种构型(type III)。我们共有1202个CDR序列,覆盖L1,L2,L3,H1和H2。其中,有600个(50%)属于I型CDR长度,有522个(43%)属于II型CDR长度。 III型CDR长度只有80个(7%)。并非我们所有的I型和II型CDR长度都是100%可预测的,因为对于这些定义,我们只需要至少85%的可预测性即可。同时,基于基因来源或序列,大多数III型CDR长度似乎是可预测的。但是,由于数量少,我们不能确定在给定其他结构的情况下这是否会成立。因此,我们估计基于基因来源(小鼠或人; k 或λ等),可以容易地预测出至少85%的非H3 CDR结构,以及CDR的序列或某些框架位置的同一性。
对于某些环长度而言,仍然存在一些挑战,这些环长度的结构和序列高度可变,目前没有明显的模式。我们的某些聚类具有较高的方差,可能表明需要将集合划分为更大数量的聚类(在亲和力传播算法中具有较低的自相似性)。无论有没有该步骤,都将需要开发从簇中为目标抗体序列选择最佳环的方法。
我们的第二个问题是“随着时间的推移,Chothia对1980年代和1990年代的分析是否成立?” Chothia等人提供了循环的20个CDRSlength组合的规范类,但H3除外。在我们的分析中,我们总共有28个CDR长度,尽管Chothia等人的许多CDR长度在PDB中没有,甚至到目前为止也很少。 Chothia分析中的20个CDR长度中,只有四个具有一个以上的经典类别。由于这四个CDR长度中的三个是小鼠/人类(L1-14)或k/λ(L1-11),或取决于脯氨酸残基的存在和位置(L3-9),因此相对容易识别。 H2-10具有一些重要的结构确定残基,包括骨架VH残基71。因此,要想利用Chothia规范类进行结构预测,需要一组复杂的结构确定残基的想法在很大程度上是不正确的,因为它们几乎全部可以仅基于CDR,环长度和基因来源来区分不同的类(L3-9和H2-10除外),并且基于某些位置上脯氨酸的存在,L3-9很容易预测。对于L1-14和L1-11,我们具有与Chothia规范类相同的聚类。对于L3-9和H2-10,我们拥有比规范类更多的集群。
虽然Chothia只有4个CDR长度且具有一个以上的典范类别,而16个CDR长度却只有一个类别,但我们有15个CDR长度且具有一个以上簇,而13个CDR长度却仅有一个簇。 Chothia等共有25个规范类,我们有72个类在一类的16个Chothia CDR长度中,我们现在将其中的10个分成多个簇。从这个意义上讲,Chothia分析并不能阻止结构数量的大量增加。
抗体为蛋白质结构预测方法的开发提供了独特的机会。尤其是,loop建模是一个具有挑战性的问题,六个不同CDR中每个CDR的300多个loop的结构提供了一个独特的数据集,用于在高度相似的核心结构的上下文中定义sequence-structure关系。虽然通常的做法是在loop建模中从多个模板中借用信息[30,31],但在其他具有许多结构的蛋白质家族中,类似的聚类方法也可能有效。
四、材料和方法
4.1 重链和轻链V域的隐马尔可夫模型
PSI-BLAST用于搜索PDB中所有序列的数据库(非冗余序列文件pdbaanr可在我们的PISCES网站上找到),使用PDB条目1Q9R中抗体结构的可变域区域。仅上述序列保留35%的同一性和优于1.0* 10^(-20)的E值,使得仅保留抗体结构域(例如,排除T细胞受体和其他Ig序列)。使用PISCES服务器以90%的同一性剔除所得的重链和轻链序列。重链序列和轻链序列的多序列比对分别通过CLUSTAL W确定,然后手动剔除和编辑。然后使用HMMER程序将这些比对用于创建重链特异性和轻链特异性隐性Markov模型。Profile HMM是蛋白质家族的多序列比对的统计模型,包括位置特异性插入概率。这使得它们非常适合于确定CDR的位置,所述CDR发生在可变结构域序列内的明确定义的位置并且长度变化。
这些HMMs用于搜索PISCES服务器上可用的pdbaa(PDB中所有蛋白质序列的集合,包括冗余)。选择HMMER得分和E值的临界值,以便在搜索pdbaa蛋白序列时,只有抗体重链和轻链序列的得分比临界值更好。由两个HMMs发现的序列被分配给得分较高而E值较小的序列。两者k,λ轻链的分数比轻链H的截止分数更好。这些图谱HMMs–一个用于重链,一个用于k轻链T–步用于鉴定每个CDR之前和之后的特定保守框架位置。 铁汉 15:46:08
4.2 定义CDRs
这项研究需要CDR的一致定义。 Kabat等仅根据抗体序列信息得出的CDR定义和残基编号方案。Chothia和Lesk和Chothia等人从抗体的最早结构中定义了CDR,并在研究规范CDR构象时使用了该方案(有一些变化)。 Martin等[39]提出了Chothia CDR定义的修改版本,该定义已在其SACS数据库中使用。Honegger和Pluckthun对16条可变链进行了多结构比对,并分析了Cα位点的变异,为了VH和VL链以及T细胞受体α,β, γ和 δ链定义一致的编号方案。他们没有严格定义CDR的边界。
在定义CDR的边界位置时,我们考虑了几个标准。
- 首先,我们想要在抗体之间几乎没有结构变异的位置。
- 其次,在可能的情况下,我们希望在β-sheet框架中彼此相对(即,将相同长度的长度延伸到框架中)。
- 第三,我们想要在VH域和VL域之间或多或少对称的定义。
图1显示了我们选择作为CDR与周围框架区域之间边界的位置的序列徽标。
L3的Kabat,Chothia和Martin CDR从形成链内一端的Cys残基开始一个残基开始。 39该位置也是Honegger和Pl-ckthun数据中CDR3变异小于0.5Å的最后一个位置。因此,我们使用二硫键第二个Cys之后的残基开始CDR L3和H3。为了鉴定CDR3的C末端,我们检查了结构并选择从Cys + 1对面的残基作为CDR的最后残基。跟随该位置的构架残基也是该构架区域中第一个在Honegger-Pluckthun数据中具有小于0.5 Å变异性的区域。这些位置在轻链和重链序列中都易于在视觉上识别。 VH链中CDR H3后面的基序几乎总是WG [X] G,其中X通常是Q,E,K,H或P。VL链中CDR L3后面的基序几乎总是FG [X ] G,其中X是G,A,S,Q或T。
由于二硫键的一端已被定义为CDR3s之前的边界残基,我们决定将H1和L1边界置于二硫键的第一个Cys与紧随其后的残基之间。这也是Kabat等人(Chothia)和Martin和Thornton对L1的定义,尽管它们的H1定义开始于5个残基(Chothia和artinSThornton)或8个残基(Kabat等)之后。值得注意的是,在此,Cys残基也是由Honegger和Pluckthun鉴定的具有a <0.5 Å可变性的最后残基。与H3和L3一样,我们将H1和L1 CDR的末端定义为与Cys + 1相邻的链中紧邻的残基。在VH和VL中,这是一个高度保守的色氨酸之前的位置。 CDR的最后一个残基和CDR之后的第一个残基在Cα位置(0.5 Å)均具有低变异性,而CDR中的残基具有高变异性。 H1之后的motif是W [VIF] [RK] [QK],而L1之后的motif通常是W [YFLV] [QL] [QEH]。
Chothia和Martin-Thornton将H2的第一个残基定义为在前面的β-折叠链中的氨基酸疏水段之后,通常具有序列LLI或WIG。该第一个残基也位于与我们上面定义的H1的最后一个残基相邻的β-折叠链中,从而使H2(和L2)的第一个残基的定义在β-折叠中对称。有了这个定义,H2或L2之前的骨架最后一个残基也是该区域中的最后一个残基,Cα位置的变异性较低。
H2和L2的C末端有些难以定义。 H2和L2在β-折叠中连接相邻的链,因此我们可以定义与起始残基相对的loop末端。确实,这对于H2效果很好,因为该段是短的β-sheet链,与该区域中通常为全线圈的VL链相反。这条短β-链的存在和位置都非常保守,其后的残基是非常保守的Tyr残基(有时为Phe),与另一个β-折叠堆积在一起。因此,我们决定在这个保守的Tyr之前,让H2以两个β-sheet残基结尾。 H2之后的序列基序为[YF] [NAVSG] [PEQD] [KDS]。此处缺乏强烈的序列共有性与Honegger-P1uckthun数据中较高的结构变异性有关,但变异性直到进入下一strand(和sheet),)才回到最小值,然后回到抗原结合位点。
但是,对于L2,该区域不是β-链,并且其后的区域显示出相当大的结构变异性。 Martin,Kabat等人和Chothia均使L2定义比上述H2定义长三个残基。该序列也是相当可变的,因此似乎有理由对L2 /框架边界相对于H2进行非对称定义。在我们的定义中,L2区几乎总是长8个残基,而Martin / Kabat / Chothia的定义是在一个残基以后开始的,因此长7个残基。 L2之后的序列基序通常是G [VI] P [SA]。根据我们的定义,CDR环路如图2所示。
在HMMs中识别出CDR之前和之后的残基匹配状态。因此,通过确定查询序列与HMM的匹配可以容易地通过确定序列中哪些残基与用这些边界鉴定的匹配状态比对来鉴定CDR边界。我们将定义与2009年SACS数据库的结果进行了比较(自更新以来)。目视检查结构中的所有差异(考虑CDR定义的差异),HMMs正确识别了我们定义的CDR位置。 SACS数据库有时将CDR中的Cys和Trp残基识别为框架的保守Cys和Trp残基,因此不一致地定义了CDR。 SACS还缺少包含不同抗体或独特型及其抗独特型的单链抗体和PDB条目的某些CDR。
4.3 筛选数据以查找定义不明确的loop构象。
在PDB条目级别,从数据库中删除了非X射线结构和分辨率低于2.8 Å的结构。应用了几个标准以从数据集中删除特定的CDR环。
- 首先,消除了缺少骨架原子的任何环。
- 其次,还将具有B因子为80或更高的主链原子的任何环也排除在数据库之外,以除去高度移动的环。我们还删除了那些缺少B因子(B = 0)的因子。
- 第三,也排除了脯氨酸以外的任何带有顺式肽键的环。我们只发现了12种这样的抗体结构。虽然确实存在非Pro顺式残基,但它们非常罕见,至少在某些情况下很可能是不良结构测定的结果。
- 第四,我们使用了最近开发的Ramachandran概率密度集,以删除任何具有极不可能的骨架构象的环。这些Ramachandran分布取决于序列,对于给定的残基类型及其在右边或右边的邻居具有不同的分布。这些可以简单地组合成残基序列三元组的概率分布。在高分辨率结构中的loop调查中,每个残基的98% loop的能量低于9.5(以任意单位来自-log(p))。
因此,我们删除了少数具有很高构象能量的环。所得的结构集是高度冗余的,在PDB中具有相同抗体的不同晶体结构的许多示例。考虑到这一点,我们比较了六个CDR的序列。对于具有相同CDR序列集的任何结构集,我们使用的是最高分辨率的结构。表1显示了筛选出不良结构和冗余步骤的效果。 铁汉 15:46:16
4.4 对每个CDR长度的聚类loop构象进行聚类
从劣质结构中挑选出数据集,我们按以下结构对聚类进行聚类。
- 首先,对于每个loop长度,我们检查了loop每个位置的二面角差异。loop通过cis-trans配置聚类。例如,L3-9(CDR L3,长度9)环通常在一个或两个位置具有顺式脯氨酸。在这种情况下,L3-9-allT(全反式)环与L3-9-cis7(在位置7的顺式Pro)和L3-9-cis6,7(在位置6和7的顺式Pro)一起分组。我们删除了少数异常值,这些异常值定义为一个循环,该循环中CDR-length组和cis-trans配置中的Φ,Ψ至少距离其他循环90度以上。
一旦按CDR,长度和cis-trans配置对环进行了分类,为了按结构将环聚类,我们需要任意两个环结构之间的距离函数。我们选择使用方向统计中使用的度量来计算两个角度之间的距离。对于两个类型相同且在两个不同结构的相同残基位置的两个二面角θ1和θ2,它们之间的距离定义为:


H3环段的广泛结构多样性使其难以聚类。特别地,它们的环区的末端在长度,序列和构象上变化很大。但是,如果将长度超过10个残基的H3区域拆分为torso区域,该区域在H3环的N端和C端与框架相连,而head区域则对应于环的尖端,则torso区域构象变量较小,并且可能符合离散的构象集。
一旦完成聚类,将合并结构彼此相似的聚类。对于具有相同环类型,长度和cis-trans配置的每对集群,计算中间环之间的构象距离(请参见等式(3)),然后除以环的长度。我们颠倒等式(2)计算每个残差的平均绝对角度差。如果每个二面角对的距离小于相差65度的两个角度的距离,则将两个簇合并。根据经验,该值在删除显然不与其他聚类的构象时效果很好。
4.5 构象类定义
为了促进不同聚类之间的比较,我们基于Ramachandran图的划分区域分配了残基构象。不同的构象类别包括A(α-螺旋区),B(β-折叠区),P(对于聚脯氨酸II),L(左旋螺旋区),D(δ)和G(γ)。这些在图4中用定义显示。然后,使用这些类基于loop的结构对群集进行批注,其中到该群集中所有其他loop的中位距离最低。此步骤提供了一种比较不同群集的简便方法。
参考资料
- North, B., Lehmann, A., & Dunbrack, R. L. (2011). A New Clustering of Antibody CDR Loop Conformations. Journal of Molecular Biology, 406(2), 228–256. doi:10.1016/j.jmb.2010.10.030
