【4.3.1】蛋白质药物的免疫原性( Immunogenicity of Protein Pharmaceuticals )
- 蛋白质疗法通过提供高度特异性且缺乏许多脱靶毒性的方案,彻底改变了许多疾病的治疗方案。
- 许多治疗性蛋白质的临床应用已经被蛋白质的不需要的免疫应答的潜在发展所破坏,限制了它们的功效并对其安全性产生负面影响。
- 本综述旨在概述治疗性蛋白的免疫原性,包括免疫机制和影响免疫原性的因素,免疫原性的影响,临床前筛选方法以及降低免疫原性的策略。
缩略词表:
- ADA, antidrug antibody;
- ADCC, antibody-dependent cellular toxicity;
- APCs, antigen-presenting cells;
- CDC, complement-dependent cytotoxicity;
- CFSE, carboxyfluorescein succinimidyl ester;
- Epo, erythropoietin;
- FVIII, factor VIII;
- HA, hemophilia A;
- IFN, interferon;
- IL-3, interleukin-3;
- ITI, immune tolerance induction;
- Mab, monoclonal antibodies;
- MHC, major histocompatibility complex;
- MTX, methotrexate;
- NSG, NOD scid gamma;
- PBMC, peripheral blood mononuclear cell;
- PK/PD, pharmacokinetic and pharmacodynamic;
- rhGAA, recombinant human acid alpha-glucosidase; RTX, rituximab;
- tPA, tissue plasminogen activator.
一、前言
蛋白质药物是增长最快的一类药物分子之一,包括临床上用于各种适应症的250多种蛋白质。重组技术的进步不仅为蛋白质的无限供应铺平了道路,而且通过消除从天然来源分离的蛋白质可能发生的病毒传播,改善了安全性。此外,通过识别影响疾病严重程度或进展的关键蛋白质并利用其治疗潜力来提供治疗,在分子水平上了解疾病有助于基于蛋白质的疗法的发展。这些例子包括分别用重组人酸性α-葡萄糖苷酶(rhGAA)和因子VIII(factor VIII , FVIII)处理Pompe病和血友病A(HA)的救命替代疗法,以及基于细胞因子的疗法,如干扰素β。另外,基于单克隆抗体的药物如阿达木单抗(adalimumab,Humira),Fc-融合蛋白如依那西普(etanercept,Enbrel)用于类风湿性关节炎,Eloctate(Fc-FVIII)和免疫检查点抑制剂如pembrolizumab(已经开发了Keytruda)和atezolizumab(Tecentriq),以及其他几种抗体产品。
不幸的是,许多治疗性蛋白质的临床效用已经被针对蛋白质的不需要的潜在的免疫应答所破坏。 几种临床批准的产品的免疫原性发生率已有很好的记录6-8(表1和表2),有些在甚至达到临床试验之前就已经失败。 抗体的可开发性可能限制功效并对安全性产生负面影响,从而妨碍蛋白质的临床应用。
1.1 免疫原性机制
1.1.1 免疫反应
由于2种不同的机制,可以产生针对治疗性蛋白质的免疫应答:经典免疫应答或通过破坏耐受性。自身蛋白质和非自身蛋白质的免疫区分是决定免疫应答机制的关键。在健康个体中,免疫系统通过胸腺中自身反应性免疫细胞的负选择来维持自身蛋白质和非自身蛋白质之间的稳态,被认为是外来的蛋白质会引发患者的经典免疫反应。该反应的特征在于抗体的形成,并且通常在施用后数天至数周内首次出现,并且通常在单次注射后触发。一旦记忆B细胞产生,这些类型的反应是持久的并且很难逆转。随后暴露于蛋白质时,免疫系统产生“二次反应”,主要表征通过显着的IgG释放,极大地影响治疗。引起经典免疫反应的最着名的治疗性蛋白质实例是替代疗法,如rhGAA18和FVIII 。它还影响单克隆抗体(Mab)治疗,因为互补决定区具有高度免疫原性和结果由于缺乏对该区域的中心耐受性而产生抗独特型同种异体抗体。与内源性自身蛋白质同源的治疗性蛋白质由于已建立的免疫耐受性而通常不会产生反应,但可通过破坏B而变得具有免疫原性。 细胞耐受性通过负选择自身反应性B细胞介导的骨髓中维持B细胞耐受,并且高BCR亲和细胞被迫进行细胞凋亡。通过重复给药,耐受性可以分解为治疗性蛋白质, 例如干扰素(IFN)-γ,IFN-β,促红细胞生成素(Epo)。
1.1.2 免疫原性的细胞和分子机制
导致抗药性抗体(antidrug antibody,ADA)形成治疗性蛋白质的细胞机制涉及2种主要细胞类型,抗原呈递细胞(APC),包括树突细胞和巨噬细胞,以及T和B淋巴细胞。由于APC的高吞噬能力,例如未成熟的树突状细胞,所施用的蛋白质被吞噬,加工,并在主要组织相容性复合物(MHC-II)的背景下呈递给淋巴结中的T淋巴细胞。该过程还伴随着APC通过上调成熟而成熟。共刺激标志物如CD40,CD80和CD86以及APC向局部淋巴结的迁移。在分泌细胞因子的帮助下,APCs刺激抗原特异性T淋巴细胞,促进B淋巴细胞活化并促进其分化为记忆B-细胞和浆细胞(图1)。记忆B细胞处于休眠状态,直到随后暴露于治疗性蛋白质,浆细胞分泌识别特定表位的抗体对APC MHC受体呈递的蛋白质的研究。通过T细胞非依赖性过程也可以实现免疫原性,其中抗原直接与B细胞结合。
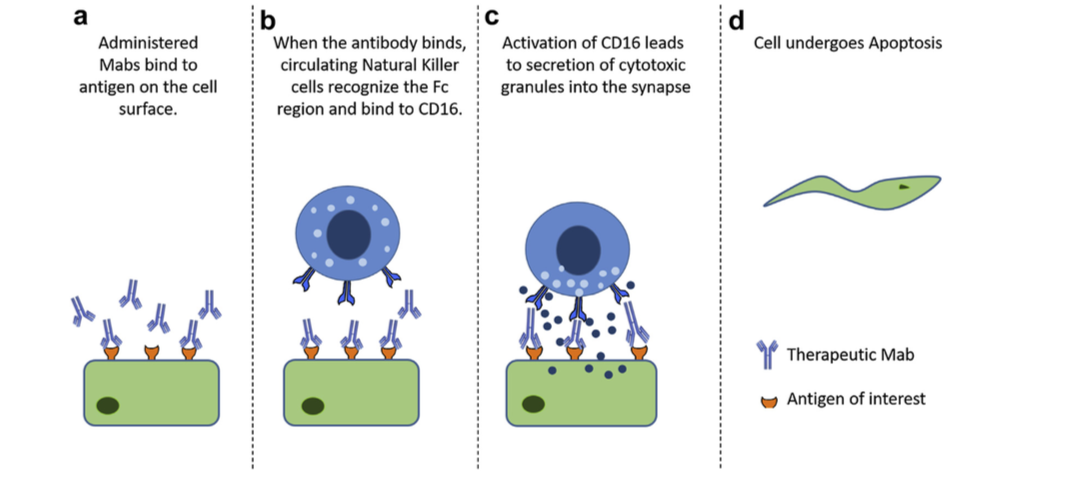
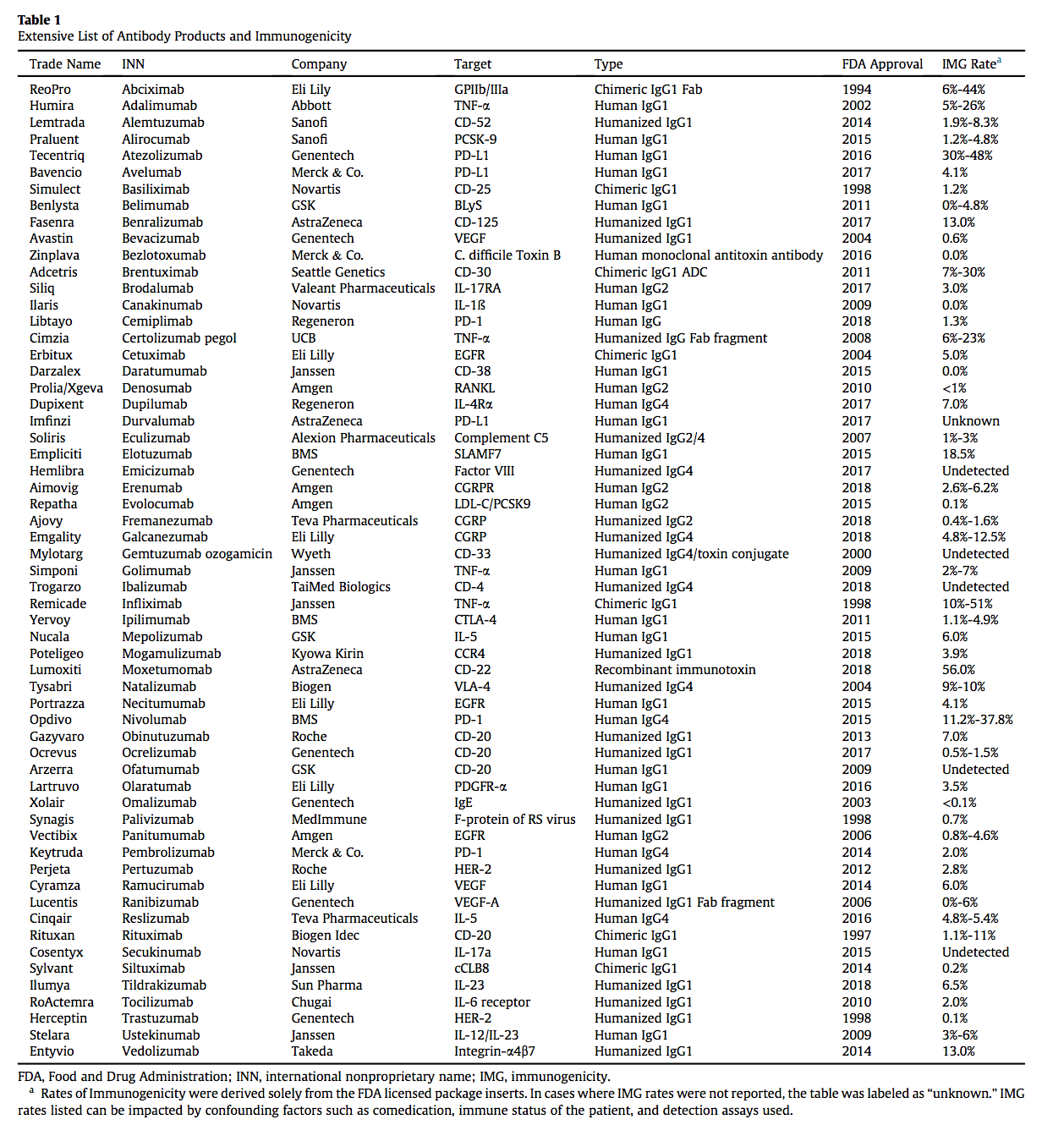
图1.抗体依赖性细胞毒性机制:当Mabs与细胞表面上的靶抗原结合时,它可导致自然杀伤细胞通过CD16与Fc区结合。 该受体的活化导致细胞毒性颗粒的分泌,其进入健康细胞,迫使其经历细胞凋亡。 (a)施用的单克隆抗体与细胞表面上的目标抗原全身结合。 (b)当抗体结合时,循环的自然杀伤细胞识别Fc区并与CD16结合。 (c)CD16的活化导致细胞毒性颗粒分泌到突触中然后进入细胞,(d)迫使细胞经历细胞凋亡(apoptosis)。
1.1.3 治疗性蛋白质和抗体的免疫应答
治疗性蛋白的免疫原性表现为ADA和超敏反应的发展。超敏(hypersensitivity)反应可根据反应机制和效应分子的产生区分为4种类型:
- I型超敏反应由IgE抗体介导交联蛋白和肥大细胞释放血管活性介质,最显着的是组胺,通常表现为过敏反应,鼻炎,荨麻疹和哮喘。在蛋白质给药后数分钟至数小时内观察到I型反应,通常可以通过预防性给予抗组胺药来预防。 I型反应也可以独立于IgE释放而发生,涉及T细胞,更常见于食物蛋白.
- 在II型反应中,针对细胞表面抗原的IgG和IgM抗体介导细胞破坏和补体激活。类型的常见例子II超敏反应是血细胞不相容。由于细胞表面标志物,来自不同类型(A,B,O)的血细胞在输血时被不同地感知,并且身体通过攻击“入侵”细胞而作出反应。用靶向细胞表面抗原的Mab产物也观察到II型超敏反应。在施用的Mab与靶或非靶组织上表达的细胞表面抗原结合后,免疫系统可引发抗体依赖性细胞毒性(ADCC),引起细胞裂解和死亡(图1)。例如,使用曲妥珠单抗(抗HER-2)观察到的心脏毒性至少部分归因于除了ADCC之外的癌组织中HER-2在心肌上的表达。与抗原结合的给药抗体可以通过结合C1q和靶细胞表面的偶然沉积,引起补体激活,导致补体依赖性细胞毒性(CDC)(图2)。抗体和抗体亚型的Fc部分的设计也可以影响细胞的细胞毒性,因为与IgG1或IgG3相比,IgG2和IgG4显示较低的ADCC,而IgG4显示没有CDC效应。
- III型超敏反应主要表征为免疫复合物介导的超敏反应。如果蛋白质的量显着高于存在的ADA,则可形成小的免疫复合物,其不被吞噬细胞有效清除,由于免疫复合物或蛋白质 - 抗体复合物沉积在各种组织中而导致III型反应。复杂的沉积可导致补体激活和引起血清病的炎症反应。ADA和蛋白免疫复合物的大小是III型反应的主要决定因素。
- IV型超敏反应(迟发型超敏反应)是细胞介导的,其特征是致敏的T辅助细胞释放细胞因子并激活巨噬细胞和T细胞。
图2.补体依赖性细胞毒性( Complement-dependent cytotoxicity ):当Mabs与靶抗原结合时,它可以沉积在健康组织中。 循环补体可以与免疫复合物结合并触发补体激活,导致细胞死亡。
根据临床观察,大约63%的针对Mab产生的超敏反应是I型反应,13%经历细胞因子释放,而约3%是延迟IV型反应。免疫相关的Mabs毒性,特别是免疫 - 调节Mabs起源于两种机制:首先,如上所述,健康组织中的靶标结合,第二,增强药理学,减弱细胞上靶分子的活性。最近,免疫检查点抑制剂(immune checkpoint inhibitor)的免疫相关不良反应被记录(反 - 细胞毒性T-淋巴细胞相关蛋白4,反编程死亡配体1和反编程细胞死亡蛋白1),其作为涉及多器官系统的炎症,包括胃肠道,内分泌系统,皮肤和肝脏,并且,在较小程度上,中枢神经系统,心血管和肺系统。这些免疫相关的不良反应可以在治疗开始后的最初几周内的任何时间出现,直至治疗停止后数月。毒性的病理生理学尚未完全了解;然而,它可能与免疫检查点在维持免疫稳态中的作用有关。目前,对于已发生这些不良免疫相关反应的患者,建议使用类固醇和免疫抑制治疗
1.1.4 免疫原性对疗效的影响
ADA对治疗性蛋白质的反应可分为2个不同的类别:中和和结合抗体。中和抗体识别治疗性蛋白质中对其生物活性至关重要的区域,直接消除其活性。例如,大约30%的重组FVIII作为替代品的重症HA患者会产生被称为“抑制剂”的中和抗体,而在这些抑制剂阳性患者中,止血效率会下降。同样,89%-100%的用rhGAA治疗的Pompe病患者会产生针对rhGAA的抗体。一旦建立持续的免疫反应,rhGAA治疗的功效就会丧失,并且没有安全的临床选择。相反,结合抗体与蛋白质相互作用并且可以改变蛋白质的药代动力学,通过减少总体全身暴露间接地影响其功效。在剂量递增临床试验中,emicizumab是一种双特异性FVIII模拟物,6名患者中的一名患者发生ADA,导致蛋白质快速清除,间接影响止血效果。同样,长期使用阿达木单抗导致发育大约28%的患者患有ADA,并且与较低的阿达木单抗浓度和较差的临床结果相关。
1.1.5 免疫原性的PK和PD建模 (PK and PD Modeling of Immunogenicity)
最近,使用药代动力学和药效学(pharmacokinetic and pharmacodynamic,PK和PD)方法来评估免疫原性对功效的影响已引起相当大的兴趣。如所讨论的,结合抗体可通过改变蛋白质的PK间接地影响PD,通常通过增加清除率。越来越多的研究结合了免疫原性的影响来开发PK模型,该模型描述了抗体对治疗性蛋白质的吸收,处置,代谢和消除的影响。已经使用了一些模型设计:包括免疫原性的模型协变量,包括ADA浓度作为连续变量的模型,以及包含免疫原性作为补充清除的模型(图3)。 Xu等将免疫原性模拟为协变量,发现它对戈利木单抗清除率有显着影响。或者,ADA的作用可以模拟为单独的清除途径,导致直接从循环中消除,而不是组织中的蛋白水解降解。 Ng等开发了阿达木单抗的PK模型,将ADA作为增加清除率的因素。 Casteele等开发了一种非线性混合效应模型来描述克罗恩病患者的复方PK,并将ADA浓度建模为连续变量。 ADA也可以被建模为分类变量,如Rosario等人所做,其中ADA基于其是否存在而建模。然而,这种类型的模型没有考虑患者中不同水平的ADA。另一个考虑因素是在模型中加入滞后组件以说明ADA开发所需的时间。 Chen等人将免疫原性建模为清除途径,并包括一系列延迟隔室,抗体在进入中央隔室之前必须通过该延迟隔室。 Gomez-Mantilla发表了一篇关于将ADA纳入PK模型领域的优秀评论

图3. 2室模型(compartment)(a)描述了ADA对蛋白质PK的影响。 将蛋白质施用到中央隔室(C1)中,在那里它可以分布到组织隔室(C2)中。 CL1和CL2分别描绘了来自中央隔室和组织隔室的clearance。 左侧描绘了免疫复合物的形成。 中央区室(ADA)中的ADA可与蛋白质结合形成免疫复合物(ADAþP),可通过降解途径(CL3)直接消除,补充CL1和CL2。 第二个模型(b)具有相同的基本结构,添加了Tlag组分(ADAL),考虑了启动免疫反应所需的时间。 ADA以假设的推注剂量开始,然后在到达中央隔室之前通过一系列隔室。
很少有研究发表使用PK和PD方法整合ADA反应,这对于完全描述免疫原性对疗效的影响至关重要。 这种PK和PD整合还需要考虑ADA对治疗性蛋白质PD的直接作用。 我们最近提出了一个综合模型来定义抑制剂开发对HA中FVIII的影响,其中FVIII的未结合部分从PK分析中阐明并与直接反应模型整合以确定抑制剂对PD的影响。
1.1.6 抗药物抗体的测量
作为新药批准的要求,在临床试验中测量患者的ADA是重要的。不同的测定可以对感知的免疫原性率和严重性产生影响,因为测定必须足够敏感以检测低水平的ADA并且能够区分治疗性蛋白质和ADA。此外,在大多数情况下,表征和比较方法通常很困难,因为人类ADA标准无法确定确切的浓度。 ADA测量的挑战包括测定对ADA而非药物产品的选择性,几种测定的假阳性和阴性的存在,以及跨实验缺乏标准化使得难以比较不同测试的不同产品的免疫原性。早期的3期临床试验可能低估或高估了蛋白质产品的免疫原性,因为药物和ADA之间的检测和分化存在限制因素。分析选择是确定患者临床结果的关键,而且与阿达木单抗和肿瘤坏死因子阻滞剂相比,这一点并未得到更好的证实。 Bartelds等表明,抗阿达木单抗的ADA与患者预后直接相关,并使用放射免疫分析法估计免疫原性。 Schouwenburg等比较了抗原结合试验和pH转换抗独特型结合试验,结果表明耐药性pH转换抗独特型试验与阿达木单抗治疗的临床结果更为相关。 Bartelds,Shouwenburg和Hart发表了一项新的检测方法,用于检测抗阿达木单抗抗体,该检测显示比以前的检测更好地检测免疫原性
桥接分析(Bridging Assays)
桥接分析是工业中用于检测ADA的最常用分析之一。 在桥接免疫测定中,药物被标记,并且ADA将在2个标记的药物分子之间形成桥接,其可以通过标记检测。 这些测定法可用于检测所有抗体类别,并可用于任何样品,因为ADA将始终与标记的药物结合。 最近,Gyrolab平台已被用于开发ADA检测的结合分析。它具有自动化和体积较小等优点,可以消除昂贵的资源和人为错误的使用。
不幸的是,桥接测定存在一些局限性。 首先,血浆中的可溶性抗原可导致药物分子桥接,导致假阳性。其次,药物干扰是一个主要问题,可能导致由于药物耐受性而导致假阴性的确定,似乎降低了免疫原性的速度。 药物耐受性是样品中可存在的游离药物的量,其不干扰ADA的检测。 如果样品中存在过多的游离药物,则药物可以隐藏样品中低水平ADA的存在。 最后,在特定情况下,预先存在的抗体可能会干扰桥接免疫测定。一个例子是类风湿因子,它可以与抗体上的Fc区结合并导致假阳性
配体结合分析(Ligand-Binding Assays)
配体结合测定是常用于检测靶标结合的方法。 Shibata等比较了3种类型的配体结合试验,以根据世界卫生组织的指导方针检测抗Epo抗体。 在他们的文章中,他们比较了表面等离子共振,电化学发光和生物层干涉测量。 电化学发光是一种通过化学和电学反应测量光的方法。 生物层干涉测量法是一种测量蛋白质表面光干扰的光学方法。 如假设的那样,每种测定法检测ADA的能力不同,证实测定选择在检测ADA中非常重要。
蛋白质特异性检测 Protein-Specific Assays
在某些情况下,蛋白质特异性测定可用于测量ADA浓度。 在FVIII的情况下,Bethesda测定法可用于特异性测量BU/mL浓度的中和抗FVIII抗体,这可以更好地比较研究之间的抑制性ADA 。此外,FVIII是少数蛋白质之一 ,有一种标准可用于通过ELISA测量ADA。 ESH8是抗FVIII抗体,可用于产生标准曲线并用于测量总FVIII ADA。 Dong等人开发了一种特异于抗PEG抗体的测定法,其中生物素-PEG与磁珠缀合,并且使用检测复合物大小变化的传感器测量与珠子结合的抗PEG抗体。 先前用于抗PEG抗体的测定通常具有高背景,这在该特定方法中被消除,因为仅传感器检测到复合物。
耐药性分析
当检测到ADA时,样品中存在药物会引起问题,因此开发了耐药性测定以克服这些问题并允许更好地量化ADA。 几个实验室进行的研究比较了药物耐受试验,检测阿达木单抗ADA,观察pH转换独特型抗原结合,酸解离试验,温度转换试验和电化学发光检测。他们得出结论,所有检测都与数量相关 ADA; 然而,免疫原性的比率在51%至66%之间,许多问题来自低ADA患者。 提高这些检测的灵敏度将最好地识别ADA阳性样品。 Mikulskis等开发了一种溶液,ELISA药物耐受试验,在与其他几种方法进行比较后检测单克隆抗体的ADA。
酶联免疫吸附测定
通过用蛋白质涂覆平板并孵育样品以测量与平板结合的ADA,ELISA也可用于检测ADA。 这种方法已经在我们的实验室中与Frey方法一起用于测量抗rhGAA抗体。ELISAs的效用可能受到限制,因为并非所有ADA都有标准。 可以使用诸如倒数稀释或光密度的改进方法代替实际浓度; 然而,这使得难以比较研究之间和实验室之间的结果。
其他
除了讨论的方法之外,还使用其他不太常用的方法。 免疫PCR是桥接测定的延伸,用于具有高药物干扰的特殊情况,其中复合物用生物素标记,即使用与DNA缀合的抗生素抗体检测。使用PCR,DNA可以量化为 评估ADA水平。 免疫液相色谱 - 质谱(LC / MS)也可用于检测血浆中的ADA。使用桥接测定法鉴定人ADA,并鉴定特异性蛋白水解切割模式以将Ig分离成亚类。 在使用免疫LC / MS之前,必须通过用生物素标记药物或将过量药物加入样品中以使ADA结合饱和来富集样品。 An等人还研究了使用LC/MS检测抗体治疗的方法,该方法也可用于检测ADA。
ADA检测的局限性
在患者样本中测量ADA有两个主要的并发症。 第一个是来自药物的干扰,第二个是测定灵敏度。 来自循环药物的干扰具有使检测复杂化的能力,并且去除游离药物可以改善这些测定。 去除游离药物的最常用方法是酸解离,其富集ADA并从样品中除去药物,从而仅检测ADA。 其次,一些研究纳入了洗脱期,以允许循环药物离开系统; 然而,这在多剂量研究中是有限的。 减少药物干扰的一种简单有效的方法是通过稀释样品来降低整体药物浓度,使其不会干扰。 敏感性问题已被充分记录,并且可以存在于任何测定中。 重要的是提高灵敏度以防止可能降低整体免疫原性率的假阴性的存在。
此外,大多数实验室已经开发了自己的ADA检测方法,并且没有可靠的标准化方法。 如果没有针对每种药物的标准ADA,标准曲线生成是困难的,这使得难以比较研究之间的结果。 此外,通过许多不同的检测方法,研究可能使用不同的方法来量化相同蛋白质的ADA并不罕见。 这些问题可能存在于较早的3期临床研究中,其中测定灵敏度不高,导致大量假阴性并且难以检测ADA,降低总体免疫原性。
在讨论PK研究时,选择合适的检测方法也很重要。 当测量对免疫原性对PK的影响时,准确定量样品中的ADA和蛋白质浓度非常重要。 使用适当的分析可以影响参数,特别是在最终阶段,其中测定的灵敏度是确定浓度的关键。如果使用不同的分析,也可能难以比较研究之间的PK曲线。
1.1.7 临床管理和监测ADA
具有免疫原性的患者的临床管理非常具有挑战性,并且取决于患者和疾病的临床状况。在患有HA的患者中,在抑制剂开发后,可以使用旁路疗法,例如NovoSeven,活化的FVIIa产品,emicizumab,通过结合FIX和FX模拟FVIII的双特异性抗体,或活化的凝血酶原复合物浓缩物。为了逆转抑制剂的发展,在一段时间内使用高剂量FVIII的免疫耐受诱导(ITI)策略使免疫系统对FVIII脱敏并诱导抗原特异性耐受。确切的机制仍然是研究的主题;然而,它已经成功使用了几十年,各种给药方案已成功用于诱导ITI。不幸的是,ITI方法非常昂贵,花费近一百万美元并且对所有患者都无效。在Pompe病治疗中发展持续抗rhGAA的ADA的患者没有其他临床选择。由于情况严重,试图利妥昔单抗,甲氨蝶呤和静脉注射免疫球蛋白(后面将讨论)的免疫抑制方案非常昂贵并且全面抑制免疫系统,使患者容易感染。免疫检查点的使用如所讨论的,免疫相关不良事件(IRAE)使抑制剂复杂化。团队方法对于管理IRAE是必要的,因为它是一个复杂的过程,因为患者必须得到支持性治疗这些事件的管理,并且必须仔细监测以减少毒性。 Villadolid和Amin对IRAE的管理进行了总结。基于这些经验,很明显ADA的临床管理具有挑战性,因为大多数治疗方法复杂,费用过高,某些患者可能无效,并且可能使患者暴露于疾病之中。风险很大。预期ADA发展的预防是可取的,并且可以使用将在以下部分中讨论的低剂量免疫抑制剂或口服纳米颗粒来实现。
对于许多蛋白质治疗剂,监测ADA可以提供生命和成本节约的益处。 ADA开发的常规监测可有效改善临床结果并降低治疗成本。 Laine等使用在芬兰收集的阿达木单抗和英夫利昔单抗药物浓度和ADA开发的数据来开发一个模型,该模型显示在实际临床实践中测试患者的ADA是成本节约的。 Vincent等也提出了一种模型,其中药物水平和ADA测量可用于预测用于慢性炎性疾病的最佳生物制剂。 ABIRISK联盟对接受IFN-b和那他珠单抗治疗的患者进行了ADA的回顾性分析,发现患者的免疫原性检测可以减少ADA阳性患者的潜在数量,并减少治疗未获益的患者的金钱损失。
1.2 影响免疫原性的因素
几种因素可以影响治疗性蛋白质的免疫原性,并且可以分类为患者,产品或治疗相关因子。 治疗的持续时间,途径和给药频率都会影响蛋白质的免疫原性。人们普遍认为皮下给予治疗性蛋白质虽然更加用户友好,但比静脉给药更具免疫原性,可能是因为皮肤树突状细胞的迁移潜能。如上所述,长期慢性疗法通常具有更高的免疫原性,频繁给药也可增加免疫原性。
可影响免疫原性的患者相关因子包括患者的免疫状态和MHC受体的多态性。 MHC(或人类中的HLA)是高度多态性的,已经鉴定出具有许多不同亚基的MHC-II的几种不同等位基因,例如DP,DM,DOA,DOB,DQ和DR。 受体亚型对表位的结合亲和力不同,因此,MHC亚型的患者间差异可影响免疫应答的产生。 免疫状态也可以极大地影响患者对蛋白质的免疫反应,因为自身免疫患者对治疗性蛋白质的反应通常比免疫功能低下的患者更强烈。
实际蛋白质产物的改变可对其免疫原性潜力产生显着影响。 MHC-II识别的免疫原性表位的存在,最终产物中的聚集体,和翻译后修饰例如糖基化都影响蛋白质产物的免疫原性。聚合物和免疫原性之间的联系目前是深入研究的主题。引起免疫反应的聚集体的性质尚不清楚,因为有几种具有不同分子和生物物理特征的聚集体已被鉴定出来参与免疫系统的能力。为了更好地理解聚集和免疫原性之间的联系,已经制定了一个分类系统,用于根据大小,可逆性,结构,共价修饰和形态学组织聚集体。例如,亚可见颗粒和天然聚集体的存在已被证明可以引起强烈的免疫反应,并且由于反复暴露于免疫原性表位而可能部分同源地破坏耐受性。此外,已经显示IFN-α的天然样聚合物比免疫原性更强。缺乏天然表位的非天然聚集体和聚集体。我们小组使用FVIII进行的研究也表明,当在HA的小鼠模型中给药时,FVIII的天然样聚集体具有更高的免疫原性。除聚集外,制剂中蛋白质和聚集体的氧化也已显示出影响免疫原性。 Jiskoot等人表明氧化介导的聚集导致IFN-b的免疫原性增加。 Hermeling等也表明氧化聚集体比天然蛋白更具免疫原性。
重组蛋白质可以在细菌细胞(例如大肠杆菌)或哺乳动物细胞(例如中国仓鼠卵巢细胞)中产生。在细菌中表达的蛋白质不经历诸如糖基化的翻译后修饰,而来自哺乳动物细胞的蛋白质经历,其可导致不同的免疫原性谱。在interferon-β产品中可以看到糖基化影响的典型例子。开发的第一种产品Betaseron是在大肠杆菌细胞中生产的,不含糖基化物。 Avonex是后来开发的,采用重组DNA技术在中国仓鼠卵巢细胞中生产。在这2种产品中,Betaseron的免疫原性比Avonex高得多,分别为35%和5%。免疫原性的差异可部分归因于这些产品的糖基化模式的变化,这些变化可导致聚集。 Mab上的糖基化模式也可以影响它们在体内的毒性特征。临床上的大多数单克隆抗体是IgG类,并且在氨基酸297处含有糖基化位点,偶尔也在Fab区域中含有糖基化位点。 Mab上的岩藻糖结构减少IgG与Fc受体的结合,减少ADCC,而较少末端结合的半乳糖降低CDC活性。关于聚集和糖基化对免疫原性的影响,有几篇非常有用的综述文章。
1.3 临床前研究预测免疫原性
在评估新的生物制品时,使用临床前方法预测临床免疫原性可能很有用。 Bococizumab是一种通过抑制PCSK9来降低体内低密度脂蛋白水平的Mab,最近在III期临床试验后被推迟,理由是免疫原性发生率较高,导致Mab随着时间的推移效力降低。因为检测到不需要的免疫原性,在III期临床试验后,因子VIIa(rFVIIa)的生物工程版本被停止进一步临床开发。生物工程rFVIIa的事后分析提出修改rFVIIa上不存在的新表位,证明了严格的临床前筛查的价值。在临床前筛选期间预测此类免疫反应的能力可以减少药物磨损,并且还可以选择更好的蛋白质候选物。已经专门设计用于降低免疫原性。不幸的是,大多数临床前免疫原性模型在预测免疫原性发生率方面的能力有限,但在评估相似产品之间的相对免疫原性方面具有不可或缺的价值,排序免疫原性多种分子和制剂,以及阐明免疫反应的机制,以进一步提高我们设计较少免疫原性蛋白质的能力。体内临床前研究通常用于筛选治疗性蛋白质的免疫原性和不同产品之间的相对免疫原性。体外方法如表位mapping,HLA亲和力和T细胞增殖试验用于评估蛋白质的免疫原性潜力和识别免疫区域。已经开发了计算机程序,其关注于蛋白质三维结构的表位mapping。
1.3.1 体内免疫原性评估
一般来说,任何人类或人源化蛋白质 当施用于动物时,治疗用途将具有免疫原性; 因此,应谨慎解释结果。 然而,小鼠模型对生物医学研究做出了重要贡献,包括我们对抗原和外来蛋白的免疫反应的理解。 对于特定应用而言,易于繁殖和容纳及操纵其基因组系统是其在生物医学研究中的实用性的关键原因。
小鼠(Mouse)模型
小鼠模型已被用于预测免疫原性,比较产品之间的相对免疫原性,并阐明免疫机制。在Krishnamoorthy等人的实验中,他们比较了几种类型的FVIII产品的相对免疫原性来确定如果他们的新FVIII fc融合物的免疫原性低于目前使用的产品。发现与B-domaine缺失的FVIII或全长FVIII相比,用rFVIII-Fc处理的小鼠具有显着更低的抗体应答,这通过具有低得多的免疫原性的Eloctate(rFVIII-Fc)的临床观察证实。在Wang等人的研究中,使用125只BALC / c小鼠通过不同的给药途径比较DNA疫苗的相对免疫原性,并显示肌肉内给药的免疫原性最小。我们实验室的研究已经使用小鼠模型显示,与基于磷脂酰丝氨酸的脂质体复合的FVIII或rhGAA的施用比单独施用游离蛋白的免疫原性显着降低。具有GAA基因敲除的那些小鼠具有显着的免疫原性。用作评估rhGAA免疫原性的模型。
随着对影响免疫原性的因素越来越感兴趣,小鼠模型也已用于比较聚集产物的免疫原性。 使用HA的小鼠模型,我们的实验室显示天然聚集体比单体蛋白和非天然聚集体具有相对更高的免疫原性。 Torosantucci等人显示聚集形式的胰岛素比商业产品具有更高的免疫原性。 Braun等用IFN-α进行了一项研究,该研究表明,在聚集体形成后,该制剂变得显着更具免疫原性,这种情况发生在几乎所有市售的IFN-α产品中。
小鼠模型的另一个重要用途是提供免疫系统活动的机制见解。 可以进行过继转移和T细胞增殖研究以评估T和B细胞在蛋白质免疫原性中的作用。 由我们小组进行的过继转移研究表明CD4 + CD25 + T细胞在抑制针对FVIII的免疫应答中的作用。 将T细胞从耐受FVIII的小鼠转移到受体小鼠体内,然后表现出显着较低的ADA发展。van Beers等人最近的一项研究表明,尽管产生了抗-IFN-b抗体,但在施用IFN-b后未发生免疫记忆。临床上也已经看到由于免疫原性而转换干扰素产物的患者。 已知抗-IFN抗体具有交叉反应性; 然而,当转换产品时,患者看不到抗体水平的增加。
HLA转基因小鼠
由于生理差异,动物MHC受体不直接模拟人HLA受体。 HLA和MHC基因是一些最具多态性的基因,由于HLA / MHC表达的高主体间变异性,很难评估蛋白质的免疫原性。为了解决这个问题,开发了模仿转基因(Tg)的小鼠模型。 人类受试者并且对特定蛋白质具有耐受性。所得到的小鼠将耐受目标治疗性蛋白质,免疫原性的发展将由自身耐受性的破坏引起,而不是对外来抗原的经典免疫应答。 已经开发出用于多种治疗性蛋白质的转基因小鼠模型,例如IFN-b,人生长激素,胰岛素,和FVII。
转基因耐受小鼠的最早使用之一是研究人胰岛素和胰岛素类似物的免疫原性。 Ottesen等表明,转基因小鼠对人胰岛素具有耐受性,并且不产生抗体。 顺便提及,他们还发现增加蛋白质修饰的数量与ADA的增加相关。 比尔斯等人使用耐受人IFN-b的转基因小鼠模型来研究对IFN-b产物的免疫原性。 他们表明,Betaferon是唯一一种破坏转基因小鼠免疫耐受性的产品。 此外,它在所有测试产品中具有最高的免疫原性,这与其他有关IFN-b免疫原性的报道一致。转基因小鼠模型是有用的; 然而,该模型的开发是劳动密集型过程,必须为每种感兴趣的治疗性蛋白质开发新模型。 表3中提供了一些转基因小鼠模型。

NOD scid gamma(NSG)小鼠是高度免疫受损的并且可以被转染以在体内模型中研究人免疫系统。 NSG小鼠缺乏大多数免疫细胞:T细胞和B细胞,树突细胞,巨噬细胞和自然杀伤细胞,以及补体和细胞因子信号传导。 杰克逊实验室率先开发了NSG模型并开发了人源化NSG小鼠模型。CD34þ人工小鼠植入脐带血衍生的造血干细胞,在小鼠体内发挥功能性免疫系统,展示 正常T细胞和炎症功能。 到目前为止,NSG小鼠主要用于研究人源化小鼠模型中传染病的病理学,但正确使用这些小鼠可以提供治疗性蛋白免疫原性机制的见解。
高阶动物模型
高级动物物种可能具有更好的临床效用,用于预测免疫原性,因为人和非人灵长类动物蛋白之间的同源性更高,并且免疫机制更具相似性。恒河猴和黑猩猩已被用于检测人生长激素和组织纤溶酶原激活物(tPA)的相对免疫原性。 Zwickl等研究了人类生长激素在恒河猴中的免疫原性,发现其结果与相对免疫原性的临床观察结果相关;然而,当试图评估免疫原性的发生率时,预测能力降低了。在另一项研究中,该小组还研究了市售tPA类似物与天然猴tPA相比的免疫原性的影响。他们得出结论,抗体开发对tPA类似物的影响不大,这些低抗体水平不会影响其酶活性,这表明免疫原性不会给临床治疗带来很大风险。
除相对免疫原性外,灵长类动物模型还可用于研究新表位可能对蛋白质免疫原性的影响。 Zwickl再次使用恒河猴来研究新表位对胰岛素免疫原性的影响。 给猴子施用人胰岛素,LysPro胰岛素和猪胰岛素,并评估抗体的发展。 他们的研究结果表明,修饰的人胰岛素(LysPro胰岛素)中存在的改变不会引起单核中的新表位,因为只有1只猴子产生低水平的抗胰岛素抗体。这些结果通过临床观察证实,显示 人和LysPro胰岛素具有相似的免疫原性发生率。
犬模型也偶尔用于评估免疫原性。 在Finn等人进行的研究中,他们在血友病B的狗模型中评估了修饰因子IX(FIX)的免疫原性。他们表明即使用野生型FIX处理后也没有针对FIX-R338L的抗体形成。 在Randolph等人的另一项研究中,他们研究了重组犬Epo的施用是否在狗中具有免疫原性,并且发现没有免疫原性风险。
动物模型的局限性
尽管存在它们的用途,但由于小鼠遗传多样性的缺乏和免疫机制的基本差异,动物模型仍然具有影响其预测免疫原性的有用性的重大障碍。甚至在人类中影响免疫原性的因素尚不完全清楚,人类和动物系统之间的差异使预测变得困难。用于开发模型或研究免疫原性的小鼠品系也可对其有用性产生很大影响。 C57BL / 6小鼠和Balb / c小鼠的免疫系统差异很大,对蛋白质的刺激反应非常不同。一般来说,与C57BL相比,Balb / c小鼠具有更高的细胞因子释放和T细胞功能。而C57BL / 6小鼠通常具有较高的自然杀伤细胞活性。此外,两种小鼠品系对促炎和抗炎细胞因子的反应不同。Toll样受体中存在人和小鼠之间的差异,抗体亚群和白细胞亚群的平衡使得难以直接关联人和小鼠免疫应答。
与小鼠模型一样,高级动物物种在预测免疫原性发病率方面仍然无效。 Gunn进行了一项研究,研究白细胞介素-3(IL-3)的免疫原性,发现所有灵长类动物均形成抗体,而临床上,很少有患者形成针对IL-3的抗体。 此外,灵长类动物不是评估单克隆抗体免疫原性的有效模型。 在van Meer等人的一项研究中,他们试图评估猴子中几种已批准的mAb产品的免疫原性的发生率和影响,并将其与现有的临床数据进行比较。 他们发现,只有59%的单克隆抗体在猴子和人类之间具有相当的免疫原性,而30%过度预测,11%预测不足。
通常用于临床前免疫接种试验的实验动物在高度卫生的条件下繁殖,并且缺乏对增强免疫系统基础活性的病原体的暴露。相比之下,人类在日常生活中经常暴露于各种病原体,以保持免疫系统的良好维持。因此,生理微生物组可能不代表天然的人免疫系统。 尽管如此,在比较相对免疫原性和阐明免疫反应机制方面,临床前模型的使用是不可或缺的,这些研究在人类的伦理上不可行或不实用。
1.3.2 体外免疫原性评估
与体内测试相比,体外系统通常耗时更少且成本更低。 体外测定可用于研究免疫应答的细胞机制,鉴定蛋白质上的免疫原性表位,并评估MHC亲和力,T细胞增殖和整个蛋白质的免疫原性作用。 理论上,体外扩增可以有效地与筛选工具一起使用,以研究免疫系统响应外来抗原的机制基础,并鉴定蛋白质上存在的关键免疫原性表位。
表位映射
表位mapping是一种稳健的方法,可以通过单独分析蛋白质的肽片段来鉴定蛋白质的免疫原性表位。治疗性蛋白质被分解为重叠的氨基酸链,并且肽暴露于免疫细胞。暴露后,可通过测量细胞因子和表面已发生炎症性免疫反应的表面标志物来评估免疫原性。 Nayak等人使用这种详尽的方法来绘制rhGAA的免疫原性T细胞表位。用游离GAA蛋白免疫小鼠,然后提取脾细胞并暴露于由全长蛋白质产生的氨基酸链。使用ELISpot测定法,评估肽的IFN-g分泌,并鉴定免疫原性表位。 Hamze等人绘制了英夫利昔单抗(infliximab)和利妥昔单抗(rituximab),以鉴定CD4 T细胞表位。使用来自健康供体的淋巴细胞,他们鉴定了存在于两种抗体可变区中的9个T细胞表位,这些表位能够刺激外周血单核细胞(PBMC)分泌多种细胞因子。已经对其他蛋白质进行了表位mapping,例如FVIII和AAV2。来自完整蛋白质的表位mapping是高度劳动密集型的,并且计算机程序通常用于鉴定可能具有免疫原性以缩小表位候选物的区域。
表位扩散可能使蛋白质上免疫原性表位的预测复杂化。在典型的免疫反应期间,T细胞活化依赖于免疫显性表位的识别;然而,表位扩散导致亚优势或隐蔽表位的识别。表位扩散可能由于内吞过程改变,可变T细胞相互作用或B细胞加工突变而发生,这导致表位识别的改变。表位传播与自身免疫疾病有关,因为它可以导致持续的组织损伤,因为免疫系统识别不同的表位。在Bernard等人的一项研究中,他们表明,在用髓鞘少突胶质细胞糖蛋白免疫后,ADA靶标发生了变化,并且鉴定了新的免疫原性髓鞘少突胶质细胞糖蛋白表位。作为表位扩散的结果,随着下一波自身反应性T细胞侵入不同的表位靶标,发生连续的组织损伤。表位扩散的不可预测性在临床前模型中极难鉴定,并且虽然可以预测免疫显性表位,但是可能更难以预测随后出现的亚优势表位。
MHC结合分析
MHC结合分析可用于检测高亲和力肽,通常与表位mapping结合使用,以鉴定具有高免疫原性风险的蛋白质区域.Salvat等人描述了一种高通量方法 肽-MHC结合试验,允许使用机械化方案在单个平板上快速分析多达90种肽。 将目标肽与对照肽和可溶性MHC蛋白以不同剂量温育以评估亲和力。 更高亲和力的肽与MHC的结。ProImmune开发了一种肽结合试验,可用于提供MHC结合数据并鉴定潜在的免疫原性表位。
T-Cell依赖性检测
在被免疫原性蛋白质刺激后,T细胞增殖并释放细胞因子以发挥其作用。 通常,通过使用胸苷或荧光标记染料(如羧基荧光素琥珀酰亚胺酯(CFSE))进行放射性标记来测量T细胞增殖。CFSE是一种荧光染料,可用于评估重复的T细胞分裂和 正在迅速取代放射性标记细胞,因为它具有较少的危险和简单性。 CFSE染色还可以伴随表型染色以区分增殖T细胞的不同亚型。 Karle等人,和Rubic-Schneider等使用胸苷评估T细胞增殖,分别测定苏金单抗和Epo聚集体的免疫原性。
除增殖外,还可测量细胞因子释放以确定免疫原性。测定细胞因子活性的类型和程度可以给出关于在这些T细胞中发生的免疫应答的一般信息。通常,ELISA和ELISpot方法用于测量体外T细胞释放的IL-2和IFN-g等细胞因子。ELISpot测定是高度敏感的方法,可用于确定分泌特定细胞因子的细胞数量。 1983年由Czerkinsky等人开发,ELISpot测定比ELISA测定更敏感,并且通常在量化细胞因子反应方面更有效。细胞因子也可通过流式细胞术检测,并且可以在与T细胞的表型一起进行到T细胞应答的类型是occurring。PBMC测定法可以使用包含几个制剂在人中提供深入的潜在免疫应答未经测试分类免疫细胞的类型,更好地模仿体内免疫系统。 PBMC制剂含有几种不同的细胞类型,包括巨噬细胞,单核细胞和淋巴细胞,并且可以包括来自适应性和先天免疫系统的细胞。在Joubert等人的研究中,通过评估细胞因子分泌和T细胞增殖,使用来自健康人类供体的PBMC来评估聚集抗体的免疫原性。
先天免疫筛查
鉴于免疫反应的先天和适应性臂之间相互作用的证据越来越多,使用先天细胞系统进行体外筛选可以提高临床前筛查的预测能力。 Ahmadi等人使用了PBMC制剂,其中CD8反应性T细胞被去除以模拟先天反应,并显示体外试验与Mabs观察到的临床免疫原性之间的良好相关性。 先天性淋巴细胞(ILCs)可用作体外系统,但尚未在治疗性蛋白质的免疫原性背景下进行探索。体外先天系统细胞系转染Toll样受体与人和小鼠的组合 巨噬细胞已成功用于筛选治疗性蛋白质制剂中的杂质
B细胞表位作图 B-Cell Epitope Mapping
B细胞识别由氨基酸序列组成的不连续表位,所述氨基酸序列与蛋白质的三维构象非常接近,所述三维构象更接近地模拟天然蛋白质。 结果,B细胞表面突变的预测比预测T细胞识别的2维表位困难得多。 在B细胞表位mapping中,鉴定了蛋白质的免疫原性区域,并且可以对蛋白质进行修饰以去除这些部分。 产生抗体的B细胞可以识别2种类型的表位:结构和功能性表位。结构表位通常较大,约16-22个氨基酸,并且包含与抗体接触的蛋白质上的氨基酸。。 功能性表位较短,3-5个氨基酸链,这影响治疗性蛋白质和抗体的亲和力。
目前,预测结构表位的最准确方法是通过使用X射线晶体学来鉴定蛋白质上与抗体结合的表位。 X射线晶体成像允许识别与抗体结合的精确序列,并且还可以提供结合强度的信息。核磁共振成像也可用于鉴定结构表位而不形成晶体; 然而,它仅限于与小蛋白质和肽一起使用。 除了先前列出的优选方法之外,电子显微镜可用于检查较大抗原(例如病毒衣壳)上的结构表位。 例如,电子显微镜用于鉴定人乳头瘤病毒16衣壳表面上的结构表位,并显示抗体结合导致构象变化。
功能性表位通常通过抗原片段化,竞争性结合或修饰测试来确定。进行片段化测定以评估抗体是否与目标表位片段结合。首先,定性方法,蛋白质印迹或ELISA通常用于评估抗体是否将与表位片段结合。阳性结合相互作用表明肽片段可以是免疫原性的。竞争性结合测定可用于确定多个抗体是否可以同时结合蛋白质上的表位,提供有关治疗性蛋白质上潜在免疫原性表位数量的信息。确定特定序列是否具有免疫原性的简单方法是修饰氨基酸链并评估其与ADA的结合。修饰测试依赖于氨基酸残基的突变以评估它们对抗体与免疫原性序列结合的影响。通过突变单个氨基酸,可以鉴定有害残基,并且如果它不会消除蛋白质的活性,则可以替代它们。
体外免疫原性评估的局限性。
预测体外免疫原性的主要问题之一是试图离体复制免疫系统。免疫系统是T细胞,B细胞,树突细胞和巨噬细胞的复杂系统,这在体外系统中不易复制。很难预测二维表位和三维蛋白质结构之间的相互作用以及APC如何通过MHC消化蛋白质以呈递表位。可能存在体内免疫细胞处理蛋白质的方式与体外测定中未检测到的体外变异性的变化。合成的肽序列可能不包含对蛋白质必需的翻译后修饰,这将导致信号转导,可能导致假阴性或假阳性。即使检测到免疫原性表位,只有在它们不能被修饰的情况下才能修饰它们。干扰蛋白质活动。与许多系统一样,遗传多样性是一个难以复制的问题。来自不同遗传背景的患者来源的PBMC是可能的;然而,需要非常大的捐赠者才能充分捕获HLA多态性以进行研究。
影响免疫原性的几个众所周知的因素,例如给药途径,频率和剂量,在体外很难预测。不可预测的治疗因素也会出现,影响免疫原性,无法预测。一个例子是Eprex,一种Epo类似物,用于增加贫血患者的红细胞生长。 Eprex的初步治疗成功;然而,一些接受Eprex治疗的患者开始出现更高的红细胞再生障碍发生率。确定该问题是抗体介导的,并且用Eprex治疗变得不那么有效。初步调查表明,从预填充注射器的橡皮塞中浸出的有机化合物是最可能的免疫原性原因;然而,进一步的测试已经得出结论,制造过程中存在的钨(tungsten)可能是Eprex聚集的根本原因。在临床前测试期间,这些小因素很可能未被发现,并且几乎没有机会预测这种情况。
1.3.3 计算预测
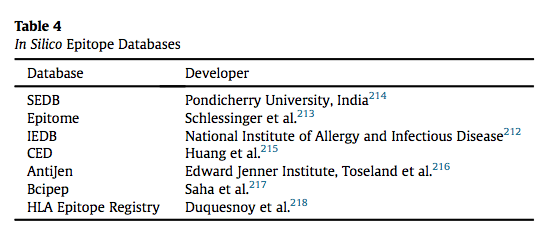
由于蛋白质组学和序列mapping的技术进步,已经构建了表位数据库,其提供了现有表位的文库和预测方法。 存在若干数据库,例如IEDB,Epitome和SEDB,其提供关于被T细胞识别的2维表位的信息。 表位和MHC之间的线性结合界面使得更容易预测,而不是受蛋白质折叠影响的B细胞表位。 表4突出显示了已开发的几个关键数据库。
还开发了几个程序和网页,可用于分析分子和预测潜在治疗蛋白的免疫原性表面。 Soria-Guerra等人回顾了几种可用于计算机表位预测的程序。 这些不同的程序包括MHC亚型预测和方法,并且一些程序能够模拟氨基酸取代以评估免疫原性的变化。 MHCPred是由Doytchinova开发的算法,可用于预测表位序列的免疫原性。 用户可以输入氨基酸序列并评估其对各种等位基因的MHC结合潜力。 EpiMatrix由EpiVax开发,用于预测治疗性蛋白质与MHC-II受体的等位基因特异性结合,并且已被改进以评估T细胞受体界面的结合。 表5总结了已经开发用于肽-MHC结合的一些算法。
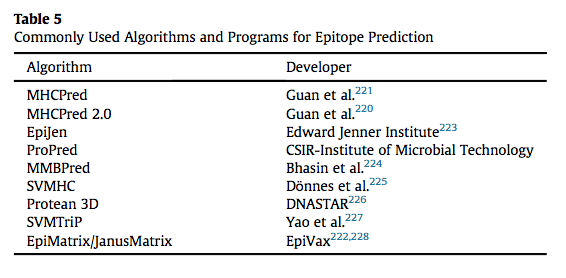
表位预测可应用于大多数治疗性蛋白质; 但是,必须结合体外方法评估预测。 预测还应与活动评估相结合,以确保改变免疫原性表位不会影响蛋白质活性。 计算机方法可以通过更大的预测肽-MHC亲和力的能力得到改善,其驱动表位免疫原性。 由于TCR识别三维结构,因此很难预测T细胞受体的肽亲和力。 如前所述,HLA多态性使得预测全人群免疫原性表达非常困难。 算法可能能够鉴定最具免疫原性的表位,但由于其个体APC呈现的替代表位,患者仍可能经历免疫原性。 翻译后的修改也难以在计算机模拟中进行。
使用定量结构活性关系模型来提高蛋白质药物的安全性和降低免疫原性,正在获得预测和减少蛋白质免疫原性区域的动力。 该方法试图使用结构,统计和基于机器的学习来将结构特征与免疫原性和活性联系起来。 从数据库,临床前和临床结果生成蛋白质结构的综合数据集可能是设计较少免疫蛋白和配方的第一步。 类似的定量结构活性关系模型已用于预测皮肤对几种化合物的敏感性。该方法也可用于预测蛋白质药物的免疫原性。 例如,Kumar等人使用计算工具使用基于结构的方法描绘表位和聚集体之间的关系
1.4 减轻治疗性蛋白质免疫原性的策略
正在制定旨在通过(1)诱导免疫耐受,(2)改变产品或(3)制剂,(4)使用小分子药物调节免疫系统来预防和逆转既定ADA反应的策略, (5)操纵T细胞和B细胞以逆转已建立的免疫反应。 已建立的免疫反应的逆转是非常困难的,通常需要淋巴细胞缺失,并且取得了有限的成功; 因此,优选诱导对治疗性蛋白质耐受的预防方案。
1.4.1 免疫耐受诱导 Immune Tolerance Induction
免疫耐受诱导是一种以抗原特异性方式预防或逆转ADA发展的方式呈递抗原的方法。免疫学上,Tregs已证明通过TGF-β介导的途径在免疫耐受诱导中发挥关键作用。我们小组正在开发一种通过将抗原与基于磷脂酰丝氨酸(PS)的脂质体共同施用,来诱导对各种治疗性蛋白质的耐受性的方法。 PS是存在于细胞内部小叶上的天然存在的磷脂。在细胞凋亡时,PS翻转到外膜并作为吞噬细胞的“吃我”(eat me)信号,吞噬和消化细胞碎片而不引发免疫反应。我们已经表明,PS与各种治疗性蛋白质和抗原(包括rhGAA,FVIII和卵白蛋白)的共同给药可以诱导耐久性耐受并阻止抗体的产生(未公开的数据)。此外,我们已经证明这种方法具有抗原特异性,因为再激发无关抗原显示出强烈的免疫应答。 Sule等人表明,用抗炎细胞因子IL-10和TGF-b调节的树突细胞能够抑制HA小鼠中的抗体发展。在他们的研究中,他们将用IL-10和TGF-β处理的树突细胞转移到HA小鼠中,然后用游离蛋白质攻击小鼠。他们还表明,该方法具有抗原特异性,因为人血清白蛋白的攻击导致正常的免疫应答。 Zhang和Scott开发了一种使用含有雷帕霉素的聚乳酸 - 共 - 乙醇酸共聚物纳米颗粒诱导对FVIII,rhGAA和AAV8的免疫耐受性的方法。这些研究表明,用其纳米颗粒施用抗原可以显着减少小鼠模型中的抗体发展。
另一种诱导免疫耐受的方法是肝基因治疗。 在该方法中,给予含有目的蛋白质基因序列的病毒载体并将其掺入肝细胞中,产生目的蛋白质。 Herzog所做的工作首次表明,腺病毒相关病毒(AAV-FIX)基因可以通过肝脏诱导免疫耐受因子IX。给予AAV-FIX的小鼠没有显示抗体发展和显着降低T- 体外细胞反应。 他们表明这种方法具有抗原特异性并诱导调节性CD4 + T细胞的增殖。 用于治疗血友病A和B的基因疗法很快引起了业界和食品和药物管理局的兴趣。
1.4.2 蛋白质修饰 Protein Modifications
通过修饰蛋白质上鉴定的免疫原性区域,直接修饰蛋白质可导致免疫原性降低。减少杂质,聚集体和宿主细胞相关蛋白进一步提高了治疗蛋白的安全性。使用蛋白质工程方法和计算机工具,已经采用去免疫和表位去除策略来设计较少免疫原性的治疗性蛋白质,其用于改造免疫原性较低的FVIII形式。 Pratt等人鉴定了FVIII蛋白上的关键免疫原性表位,并评估了可降低免疫原性的氨基酸取代,同时维持促凝血活性。使用体外T细胞增殖测定和MHC结合测定,他们确定全长FVIII的2196个氨基酸处的取代减少了T细胞增殖,同时保持了活性,说明了体外方法开发免疫原性较低的FVIII的能力。此外,Pastan等人通过鉴定和消除免疫原性T细胞表位,开发出免疫原性较低的抗CD25免疫毒素。氨基酸修饰后,他们发现T细胞反应减少了74.8%,同时维持细胞的细胞毒性。已经显示降低Mab的临床免疫原性的一种策略是增加抗体中的人类含量。一个实例显示,将抗体OKT3的构建体从鼠转变为嵌合,人源化或完全人抗体,大大降低了免疫原性的发生率。 Anne de Groot在她的综述中回顾了几种生物工程免疫原性较低的蛋白质的潜在机制。
1.4.3 与配方相关的变化 Formulation-Related Changes
修饰制剂以减少聚集也可有助于降低治疗性蛋白质免疫原性。 我们的实验室已经表明,O-磷酸-L-丝氨酸的存在通过占据在水性制剂中易于聚集的脂质结合区来防止FVIII聚集体的形成。 Senga和Honda显示在Mab制剂中包含小肽AF.2A1将结合非天然IgG构象的Fc区。 一旦结合,可以分离制剂以除去与AF.2A1结合的Mab,并且所得制剂将没有聚集体。 AF.2A1肽珠可用于除去聚集体的制剂,而O-磷酸-L-丝氨酸可用于防止FVIII制剂中聚集体的形成。
1.4.4 免疫调节药物 Immune Modulatory Drugs
小分子药物也可用于通过主要通过免疫抑制调节免疫应答来帮助患者更好地耐受治疗性蛋白质。常用的免疫抑制药物是甲氨蝶呤,雷帕霉素,硼替佐米和环磷酰胺。甲氨蝶呤(MTX)是一种抗癌药物,通常用于治疗某些类型的癌症和自身免疫性疾病,如乳腺癌,白血病,类风湿性关节炎和克罗恩病。 MTX用于治疗自身免疫疾病,因为它具有抑制T细胞活化和下调B细胞活性的能力。 Kishnani等人和Joseph等人开发了低剂量MTX方案,用于患有Pompe疾病的患者,以诱导对给予rhGAA的耐受性,并在临床上显示出预防免疫原性。 Maini等。表明MTX与抗肿瘤坏死因子抗体英夫利昔单抗的使用减少了抗体的发展,并改善了患者的反应。雷帕霉素是一种小分子大环内酯,通常通过雷帕霉素受体的机制靶标抑制T细胞和B细胞的活化来预防移植排斥。如上所述,Scott和Zhang在他们的聚乳酸 - 共 - 乙醇酸纳米粒子中使用雷帕霉素来诱导耐受性。 Herzog等使用雷帕霉素与口服递送的FVIII组合以诱导HA小鼠对FVIII的耐受性。硼替佐米是一种小分子药物,可抑制蛋白酶体活性,阻止促凋亡信号的降解,导致细胞死亡。它已被用于婴儿Pompe病患者以及MTX和利妥昔单抗(RTX),以逆转高持续性ADA并恢复治疗效果。环磷酰胺主要用作化学治疗剂以杀死快速增殖的细胞,但低剂量已被认为通过其对树突细胞稳态和细胞因子分泌的影响而具有免疫调节作用。历史上,已显示环磷酰胺在作为预处理给予时诱导对FVIII的耐受性。调查疗法还包括环磷酰胺/类固醇组合以诱导FVIII抑制阳性患者的耐受性,并且还已经测试用于预防Pompe疾病治疗中的ADA形成无效。Azathioprine是一种免疫抑制药物,可与英夫利昔单抗联合给予改善克罗恩病的治疗方法。 Colombel等人提出,改善可能部分归因于降低英夫利昔单抗的免疫原性。此外,硫唑嘌呤与英夫利昔单抗或阿达木单抗联合使用可使ADA频率降低47%。
1.4.5 淋巴细胞操作逆转ADA发展 Lymphocyte Manipulation to Reverse ADA Development
除了耐受诱导策略之外,还研究了操纵和消除淋巴细胞以预防或逆转针对治疗性蛋白质的免疫应答的方法(图4)。这些方法不仅会影响ADA的发展,还会损害患者的免疫系统,使他们容易受到感染。 Kishnani等人开发了一种免疫抑制方案,该方案显示能够阻止和逆转婴儿Pompe病患者中针对GAA的抗体(例如RTX,MTX和γ球蛋白)的发展。虽然这种方法能够阻止抗体发展,但它也导致在RTX和MTX给药过程中CD20阳性B细胞显着减少,直到用RTX和MTX治疗停止后才恢复。通过改变细胞因子的产生以促进Th1应答,抗CD3抗体已成功地用于预防小鼠中针对rhGAA和FVIII的抗体发展。然而,它们无法长期调节ADA发育,可能是由于产生了对治疗性蛋白质具有反应性的新淋巴细胞。使用免疫抑制药物不会诱发积极耐受;因此,它仅维持一个细胞寿命。

图4.各种免疫调节药物对免疫系统的影响。 雷帕霉素(Rapamycin)通过雷帕霉素途径的机制靶标抑制T细胞和B细胞活化。 利妥昔单抗(Rituximab)阻断CD20,杀死表达CD20的B细胞。 甲氨蝶呤(Methotrexate)抑制DNA合成并杀死快速分裂的细胞。 硼替佐米抑制蛋白酶体(Bortezomib inhibits proteasomes),促进快速分裂细胞的凋亡。
Scott等人设计了表达FVIII的细胞毒性T细胞,其可以选择性地靶向并杀死FVIII特异性B细胞。 他们在FVIII蛋白上设计了免疫显性表位,其在细胞毒性T细胞表面上表达为抗原受体,其在体内和体外结合并特异性地杀死FVIII反应性B细胞。 为了B细胞靶向,Brettschneider等将FVIII的C2结构域与铜绿假单胞菌的内毒素A融合,其选择性地靶向FVIII特异性记忆B细胞并消除它们。
有多种方法被评估以诱导免疫耐受,并且这些方法的临床可译性仍在研究中。 抗原特异性耐受是所需的方法,因为患者不会对继发感染敏感。 这些耐受诱导策略还需要一定程度的风险评估。 在某些情况下,例如Pompe疾病,几乎所有患者都会出现抗体,使患者无需缓解,也没有其他临床选择。 在这种情况下,给予免疫抑制方案可能是一种更可接受的选择,而在其他疾病中,免疫原性的发生率较低,患者可以转变为另一种产品。 减缓战略仍在制定中; 然而,有几种有前途的方法正在研究中。
二、结论
治疗性蛋白质的临床免疫原性是一个日益严重的问题,有可能破坏强效蛋白质药物的发展。了解免疫反应的机制和影响治疗蛋白免疫原性的因素将使我们能够设计更安全的药物并预防免疫原性。工程蛋白本质上较少的免疫原性或开发方法来预防或逆转对治疗性蛋白的既定免疫应答可以帮助解决免疫原性的主要问题。有几种方法可以尝试预测治疗性蛋白质的免疫原性,但是对于这些预测工具的有用性仍存在很大争议。如上所述,体内模型对于相对免疫原性排序最有用,但由于人和动物免疫系统之间的基本差异,免疫原性发生率的预测在模型中非常难以进行。体外模型可用于预测蛋白质是否与MHC分子相互作用;然而,这些系统不会将蛋白质暴露于可影响人类患者免疫原性的一系列因子中。计算机模型正在迅速发展,可用于预测免疫原性表位,但需要与体外试验相结合,以确保预测是有用的。尽管存在局限性,但用于预测免疫原性的临床前方法有助于药物开发。治疗性蛋白质的免疫原性是临床挑战,但克服它的成功将提高几种救生疗法的安全性和功效。正在积极寻求几种缓解策略,不仅可以预防而且可以逆转既定的免疫反应。
参考资料
- Dingman, R., & Balu-Iyer, S. V. (2019). Immunogenicity of Protein Pharmaceuticals. Journal of Pharmaceutical Sciences, 108(5), 1637–1654. https://doi.org/10.1016/j.xphs.2018.12.014
