【4.4.2】预测MHC I的process过程(MHCI-NP)
官网: http://tools.immuneepitope.org/mhcnp/
我们提出了MHC-NP,一种预测由MHC途径天然加工的肽的工具。 该方法是第二届免疫学机器学习竞赛的一部分,并获得了预测人类HLA-A * 02:01,HLA-B * 07:02,HLA-B *洗脱的肽段的最新准确性 35:01,HLA-B * 44:03,HLA-B * 53:01,HLA-B * 57:01和小鼠H2-Db和H2-Kb MHC分子。 我们简要解释了导致开发此工具的理论和动机。 预期在免疫学领域特别是基于表位的疫苗中的普遍适用性。 我们的工具可在线免费获得,并由 http://tools.immuneepeepitope.org/mhcnp/ 的免疫表位数据库托管。
一、前言
基于抗原决定簇的疫苗对于目前尚不可行或无效的现有方法(如病原体减毒)显示出广阔的前景。 这样的疫苗具有通过靶向抗原的免疫原性区域和宿主的特定MHC等位基因,而更易于产生和诱导非常特异性的免疫应答的优点(Toussaint和Kohlbacher,2009)。 用于识别免疫原性表位的计算机辅助方法代表了促进创建这些下一代疫苗的重要步骤。 理想地,这样的疫苗可以适于靶向人群的特定部分,例如孕妇和/或免疫功能低下的个体,对于它们而言,减毒疫苗可能会带来更大的风险。
主要的组织相容性复合物(MHC)负责特异性抗原识别并诱导适当的细胞应答。 MHC分子是负责在抗原呈递细胞(APC)表面呈递抗原的复杂途径的一部分。 此类抗原以MHC-肽复合物的形式存在,其中肽是抗原蛋白序列的片段。 一旦MHC–肽复合物出现在APC的表面,T细胞就会识别出特定的复合物并触发适当的免疫反应。 MHC分子分为两类,MHC I类为CD8 T细胞驱动应答的抗原,而MHC-II为CD4 T细胞应答抗原。 两种途径都是复杂的,并且肽与MHC分子的结合可以通过体外或计算机方法验证。
已经提出了许多方法来鉴定MHC-肽复合物(Zhang等人,2012; Lundegaard等人,2008;Giguère等人,2013)。这些方法大多数都集中在预测给定肽与MHC分子的结合亲和力上。不幸的是,肽和MHC分子的结合不足以确保肽将被加工到细胞表面。实际上,仅结合MHC分子的肽的子集可以被自然加工。预测此类肽是一项艰巨的任务。
为了完善表位肽鉴定方法,我们提出了一种预测肽是否由MHC途径天然加工的方法。我们的方法已经过D.K.Crockett和V.Brusic(Zhang和Brusic,2013年)提供的体内和体外数据的训练。 所提出的方法是基于统计学习的。因此,鉴于有训练数据可用,它可以用于MHC-I和MHC-II途径。另外,如果与任何结合亲和力预测工具结合使用,我们的方法可以验证肽是否可以通过MHC途径加工。在2012年免疫学机器学习竞赛(MLI)的背景下,我们获得了经验性结果,表明我们的方法将在免疫学,疫苗学和移植排斥中具有广阔的应用前景。
二、材料和方法
2.1 数据
用于训练我们方法的数据集是提供给2012年免疫学机器学习竞赛参与者的数据集。 这些数据集包含八个MHC-1分子,由六个人类分子组成(HLA-A * 02:01,HLA-B * 07:02,HLA-B * 35:01,HLA-B * 44:03,HLA-B * 53:01和HLA-B * 57:01)和两个小鼠分子(H2-Db和H2-Kb)(Zhang和Brusic,2013)。 对于每个靶分子,提供三组肽:结合肽,非结合肽和通过洗脱(elution)获得的肽。 通过洗脱获得的肽通过MHC-1途径天然加工。 我们的方法是使用一组由结合,非结合和天然加工肽组成的数据进行测试的。 该测试数据由MLI竞赛的组织者提供,并可以从 http://bio.dfci.harvard.edu/DFRMLI/HTML/natural.php 公开获得。
2.1.1 讨论区
从机器学习的角度来看(有关入门书籍,请参见Hastie等人(2001年)),训练数据在许多方面都具有挑战性。
- 首先,尽管训练示例由氨基酸序列组成,但大多数学习算法都以实值特征向量的形式接收数据。
- 第二,训练肽的长度从8至11个氨基酸不等。 大多数学习算法需要计算仅针对相同长度的向量定义的向量运算。 因此,取决于肽长度的肽的任何实值载体表示都将最终失败。
面对这样的问题时,一些作者更喜欢按长度对肽进行分组,并为每种长度定义独立的学习问题。 这种解决方案不可避免地导致准确性降低,因为这严重减少了每个问题的训练示例数量。 下一节将介绍字符串内核(string kernels)的概念,它为这两个问题提供了解决方案。
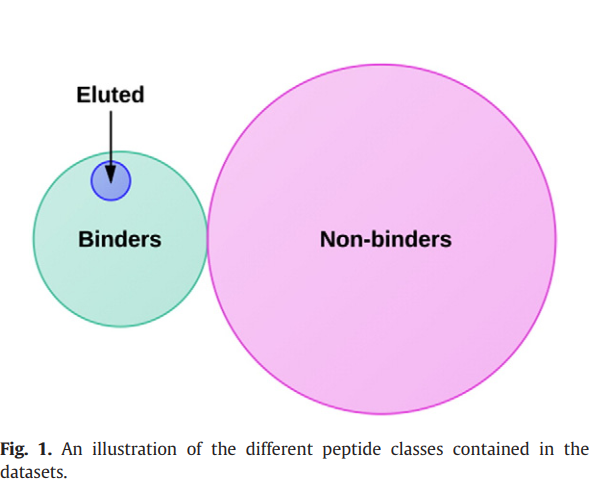
鉴定天然加工的肽的特殊情况不能描述为典型的二元或多分类问题。 实际上,在该分类方案中,每个示例可能仅属于预定义的类之一。 然而,就我们而言,天然加工的肽都可以与MHC结合,而只有5-15%的结合肽是天然加工的。 此外,由于肽可能是已知的结合物,但尚未通过洗脱鉴定,因此该问题在数据中具有固有的噪音。 这意味着洗脱的肽类别是结合肽类别的子集,如图1所示。因此,我们需要使用对噪声稳定的学习算法,并考虑这些类别之间的关系。
2.1.2 预处理数据
所提供的数据是通过实验过程获得的,因此需要进行少量的预处理以确保它不包含任何不一致之处。
- 首先,对于每个MHC分子,丢弃同时处于非结合和结合集中的肽。
- 第二,将同时洗脱和结合组中的肽从结合组中除去。
请注意,洗脱的肽也与MHC分子结合,尽管我们希望洗脱的肽有不同的集合。 这两个步骤确保了三组肽是不相交的,因此每组中包含的肽具有一个共同的特征(洗脱结合剂,非洗脱结合剂,非结合剂)。 我们将这三组肽分别称为“ E”,“ B”和“ N”。
2.2 学习方式
机器学习是面对复杂现象时构建预测软件的一种选择方法。分类是将可能有许多预定义类之一分配给由关注现象产生的每个示例的任务。在本文中,我们将限于二进制分类的情况,其中每个示例只能属于两个类。
学习算法应使任务损失最小化。在这里,任务是将天然加工的肽与所有其他肽(非洗脱结合剂和非结合剂)区分开。因此,在开发我们的方法时,我们专注于学习MHC途径天然加工的肽的预测因子。因此,我们仅考虑了以下两类:天然加工的肽(E)和非天然加工的肽(B和N)。该预测器已提交给MLI 2012竞赛,最终产生了出色的预测精度。在整个本文中,我们将此模型称为(E与BN)。
为了进一步改进此方法,自MLI竞赛以来,我们已经开发了替代模型。该模型包括学习两个预测因子:区分结合肽(E和B)和非结合肽(N)的预测子以及区分洗脱肽(E)和结合肽(B)的预测子。预测以决策树方式完成:首先应用(EB vs N)预测因子,然后,仅当预测该肽为结合剂时,才使用(E vs B)预测因子来确定该肽是否天然处理。在整个本文中,我们将此模型称为(EBvsN + EvsB)。
为了进行比较,学习了结合/非结合预测因子来区分结合肽(E + B)和非结合肽(N)。该预测因子有目的地不将天然加工的肽与结合肽区分开。这种预测因子允许与依赖MHC肽结合亲和力的当前工具建立直接比较。该预测因子还可以测量我们的方法将天然加工的肽与结合肽区分开的能力。
2.2.1 学习算法
为了学习上一节中描述的预测变量,我们使用了Cortes和Vapnik(1995)最初提出的软边际支持向量机(SVM)算法。这种监督学习算法被认为是分类问题的最新技术。
SVM算法的输入包含一对对。对于这些对中的每一个,第一个元素是一个示例,第二个元素是一个由专家赋予该示例的类。首先,每个学习示例都映射到一个可能称为特征空间的高维向量空间。然后,SVM找到一个分离的超平面,将特征空间分为两半,这样,来自同一类别的示例位于同一侧。同样,超平面使得所有学习示例都具有距其最大的欧几里得距离(也称为几何余量)。软边距SVM提供了一个参数,可以在正确分类的示例上的分类错误数量和几何边距大小之间进行权衡。这允许SVM容忍给定类的某些示例位于超平面的错误一侧,从而防止超平面受到数据噪声的影响。通常通过第3节中描述的交叉验证技术来调整此参数。
为了进行预测,首先将新的测试示例映射到与培训示例相同的空间。 然后,根据其相对于超平面的位置来分配其类别。 示例的几何余量越大,预测器对其预测类的信心就越大。 可以使用诸如普拉特定标(Platt scale)的技术将余量的概念进一步转化为概率估计(Platt,1999)。 我们建议的工具:MHC-NP,为每个新肽输出由指定MHC途径天然加工的肽的概率估计。
2.2.2 从序列到特征向量
内核技巧(Shawe-Taylor和Cristianini,2004年)是一种将复杂结构(例如字符串)映射到其中定义了点积的高维向量空间的方法。核函数是一种函数,它隐式计算此高维空间中的点积,而无需将结构显式映射到该空间。因此,这样的功能避免了在高维向量空间中计算点积的过高的计算成本。回想一下,构造超平面的每种算法都在很大程度上取决于点积运算。另外,对于点积,内核可以解释为相似性函数。两个示例越相似,内核函数值就越大。已经提出了针对多种生物学结构的内核功能,包括蛋白质一级结构(Saigo等,2004)和蛋白质三级结构(Qiu等,2007)。
许多机器学习算法(如SVM)已通过内核功能评估替换其目标函数和预测函数的点积运算而被内核化。如果存在为该数据类型定义的内核,则此类内核化算法可以处理诸如字符串之类的复杂数据。计算两个字符串之间相似度的内核称为字符串内核。毫无疑问,字符串内核是生物学中机器学习算法使用量增加的部分原因。
2.2.3 通用字符串内核 The generic string kernel
如上一节中强调的那样,字符串内核是可与内核化学习算法一起使用的相似性函数。一种常见的方法是通过选定的k值,通过其共同的k-mers比较字符串。例如,“ LFQLITA”和“ LFQRPPLI”可以分为它们的2聚体,分别是(LF,FQ,QL,LI,TA)和(LF,FQ,QR,RP,PP,PL,LI)。然后,这两个序列之间的相似性值由共同的2-聚体的数目给出。在此,相似度为3,因为序列具有LF,FQ和LI作为共同的2聚体。这种相似性函数对应于Spectrum内核(Leslie等,2002)。请注意,此内核允许比较不同长度的字符串,并对应于高维向量空间中的点积。因此,这样的内核减轻了分别考虑不同长度的肽的需要,并允许更好地使用数据。
使用k-mer比较肽是一种简单的方法,可广泛估算其相似性。但是,对于大多数蛋白质,特别是对于MHC,肽与蛋白质的相互作用发生在一个非常特定的位置,即结合位点。残基在该结合位点的相对位置及其理化性质是驱动相互作用的关键方面。尽管如此,Spectrum内核并未说明这两个重要的生物学特征。
为了考虑残基在相似性函数中的相对位置,Meinnicke等人。(2004年)提出了Oligo核( Oligo kernel),这是第一个考虑k-mers相对位置的字符串核。寡核苷酸无需计算常见k-mer的数目,而是根据它们在两种肽中的相对位置为每个常见k-mer分配权重。例如,在肽“ LFQLITA”和“ LFQRPPLI”中,2聚体“ LF”和“ FQ”具有相同的位置。相反,2个单体的“ LI”分别位于位置4和7。因此,“ LF”和“ FQ”对相似性的贡献应大于“ LI”的贡献。 Oligo内核的作者建议通过使用与肽中距离成反比的函数来加权常见k-mers的贡献。
此外,为了解释结合位点中残基的物理化学性质,Toussaint等人(J.Med.Chem。)(1992)。 (2010年)提出权衡k-mers对其理化特性的影响。这是基于这样的事实,即肽中的氨基酸有时可以被具有类似特性(例如疏水性,电荷或分子量)的另一种氨基酸取代,而不影响肽的结合亲和力。 Toussaint和合作者建议通过使用其氨基酸的理化性质来比较k-mers,从而将这些知识纳入内核功能。
受到Meinicke等人思想的启发(2004),提出了通用字符串(GS)内核。在k-mer的比较中,该核心解释了理化性质和氨基酸的相对位置。在单个目标和泛特异性肽段MHC-II结合亲和力预测基准数据集和三个定量结构亲和力模型基准数据集上,GS内核的性能优于最新的预测方法。 Giguère等。 (2013年)还提出了一种动态编程算法,用于快速计算其内核,并表明GS内核在高维向量空间中诱导了点积。 GS内核具有四个参数,即两个参数用于控制比较氨基酸的理化性质的重要性,一个参数用于设置k-mers的最大长度,另一个用于控制因k-mers的相对距离而导致的罚分。如下一节所述,可以通过交叉验证来调整这些参数。
2.2.4 实施细节
为了确保可重复性,所有实验都是使用Scikit-Learn库的SVM实现(Pedregosa等,2011)和GS内核(Giguère等,2013)进行的,可从 http://graal.ift. ulaval.ca/gs-kernel/ ,免费和开源软件。
三、理论/计算
3.1 评估模型性能
为了评估不同方法的预测准确性,我们使用了ROC曲线下的面积(AUC)和F1评分(Bradley,1997)。 此外,我们使用敏感性和特异性来分析我们方法的性能。
接收者操作特征(ROC)曲线通过绘制召回率和(1-特异性)与阈值的函数关系图,以图形方式说明了预测变量的召回率和特异性。 这种类型的曲线可用于选择关于召回和特异性之间的权衡的阈值。 AUC是通过计算ROC曲线下的面积而获得的与阈值无关的度量。 该指标与(Zhang and Brusic,2013)用于评估提交给2012 MLI竞赛的方法的性能的指标密切相关,该指标由
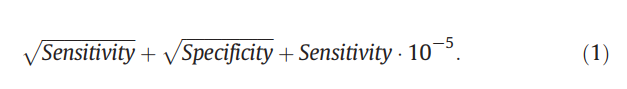
实际上,Brusic等人(2013年)通过选择最大化阈值对方法进行排名,从而使它们的度量阈值独立。
在机器学习领域,准确性是阈值相关度量,用于评估预测方法。 不幸的是,用于训练我们方法的数据集是不平衡的,这意味着洗脱肽的数量比未洗脱肽的数量小得多。 因此,使用准确性评估我们方法的性能可能会产生误导。 例如,如果只洗脱了数据集中包含的5%的肽段,则预测变量可能无法预测要洗脱的任何肽段,但准确率仍达到95%。 因此,我们建议使用F1评分来评估我们方法的判别力。 F1分数由等式给出。 (2),其值在0到1之间。此指标的优点是不受类别不平衡的影响。
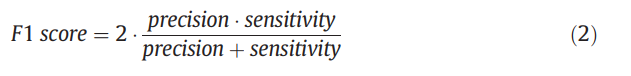
值得一提的是,决策阈值的细微变化可能会导致阈值相关指标(例如F1分数)发生重大差异。但是,比较使用训练数据估算良好阈值的方法的能力至关重要。如果没有阈值,则方法只能提供置信度分数以表明其对属于目标类的示例的置信度。虽然,对于大多数实际应用而言,必须做出二进制决定。因此,需要一个阈值来区分肯定的决定和否定的决定。此外,最近的学习范式,例如转导学习(Joachims,1999),领域适应(Jiang,2008)以及积极和无标签的学习(Elkan和Noto,2008)旨在通过使用大量的无标签数据专门为目标任务设计学习模型。 。当很少有标记的培训示例可用时,这些新方法已被证明优于监督学习。仅使用阈值无关的指标可能无法检测到此类方法的其他判别能力。为了确保可以识别将来方法的改进,我们建议使用与阈值相关的指标,例如F1分数。
3.2 算法参数选择
对于所有模型,训练集上的10倍交叉验证(请参见Hastie等,2001)用于选择SVM和GS内核参数。 结果和讨论部分报告的所有度量标准值都是根据2012年MLI竞赛组织者提供的(独立)测试集计算得出的。
3.3 高级参数调整
在交叉验证方法中,选择单个参数以产生具有有限数据量的良好预测变量。该参数是根据算法产生准确预测变量的能力估算的。但是,由于数据量有限,估计中的不确定性告诉我们,还应考虑使用其他参数值获得的预测变量。在这种情况下,贝叶斯理论建议使用具有不同参数值的许多预测变量的(概率)组合(Lacoste等人,未发表)。这样的组合允许在确定哪个预测变量真正最准确时考虑不确定性。详细的重复重采样技术可以估算每个预测变量最准确的概率。然后,这些概率将用于在最终决策中对每个预测变量的贡献进行加权。
在MLI的背景下,使用了该方法的初步版本,对HLA-B * 07:02,H2-Db和H2-Kb等位基因产生了出色的结果。但是,其他实验表明,这种方法不能显着提高AUC或F1分数。
四、结果和讨论
4.1 选择用于洗脱肽预测的模型
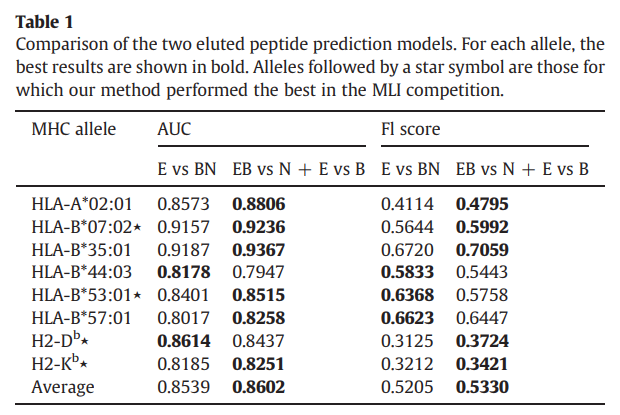
在本文中,我们介绍了两种洗脱的肽预测模型:一种预测方法(E vs BN)和两种预测方法(EB vs N + E vs B)。为了确定哪个模型最准确,我们在每个等位基因的测试集上计算了它们的AUC和F1分数。所有八个等位基因的结果显示在表1中。对于AUC和F1评分结果,我们观察到(EB vs N + E vs B)方法优于(E vs BN)八个等位基因中的六个。不幸的是,等位基因的数量不足以计算p值,因此未对结果进行统计分析。尽管如此,(EB vs N + E vs B)方法显然比(E vs BN)方法具有更好的预测准确性。
从机器学习的角度来看,此结果很有趣,因为它表明最好分别处理结合亲和力预测任务和洗脱的肽预测任务。
两种方法都具有通过交叉验证调整的参数,以非常适合预测任务。在最重要的参数中,有内核函数的参数,它们有效地定义了专门的相似性函数。通常,这些参数根据学习任务而变化很大。
对两种预测方法(EB与N + E与B)进行了两次调整,一次用于区分结合非结合肽,第二次用于分离洗脱的肽。 为每个任务找到的最佳参数差异很大。 由于两个任务明显不同,因此需要不同的相似性功能,因此可以得到此结果。 我们认为,这是两个预测器方法优于单一预测器方法的主要原因之一。 另外,请注意,前者通过考虑洗脱的肽也是结合物来利用问题的结构。
由于其优越的准确性,我们选择使用(EB vs N + E vs B)方法来完善我们的洗脱肽预测工具MHC-NP。 该工具由免疫表位数据库(Peters等,2005)托管,可从 http://tools.immuneepeepitope.org/mhcnp/ 公开获得。
4.2 与结合亲和力预测方法的比较
2012年免疫学机器学习竞赛的目的是评估预测MHC-1途径天然加工的肽段的计算方法的能力。已知肽的结合与其免疫原性之间存在相关性(Toussaint and Kohlbacher,2009)。因此,强结合剂更可能通过MHC途径自然加工。因此,我们选择比较最准确的洗脱肽预测因子(EB对N + E对B)与两种最新的MHC-1结合亲和力预测方法。第一种方法称为(EB vs N),其灵感来自Giguère等人的肽-蛋白质结合亲和力研究。 (2013)。请注意,此方法等效于执行(EB vs N + E + B)方法的第一个预测任务。该比较允许估计将(E vs B)预测因子与结合亲和力预测因子结合使用后产生的附加判别力。为了完整起见,我们还将我们的方法与流行的NetMHC-3.2(Lundegaard等人,2008)进行了比较,该方法在2012年MLI竞赛中被用作基准方法。
表2显示了三种方法在测试仪上的AUC结果。我们的方法在八个等位基因中有五个优于(EB vs N)方法和NetMHC-3.2。此外,MHC-NP的平均AUC达到0.8602,这比两种结合亲和力预测方法分别获得的0.8454和0.8265都要大。
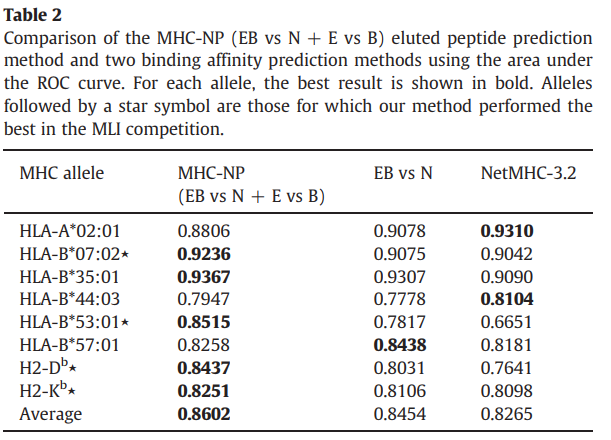
如表3所示,我们的方法在八个等位基因中有五个优于EB vs N方法和NetMHC-3.2。与EB vs N和NetMHC-3.2方法分别获得的0.4983和0.4324相比,我们的方法还获得了最佳的平均F1得分:0.5330。
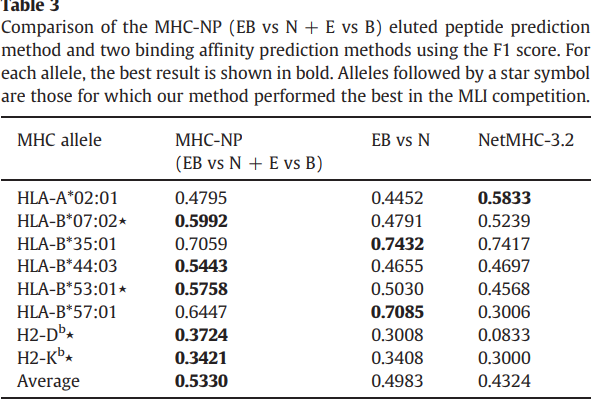
考虑到我们的结果,目前尚不清楚为什么在某些等位基因上,更简单的预测MHC-肽结合亲和力的方法在预测洗脱肽方面优于MHC-NP方法。 2012年MLI竞赛的一部分是评估后一项任务是否可以学习。我们的结果表明,对于大多数等位基因而言,可以很好地学习该任务。 HLA-A * 02:01等位基因的NetMHC-3.2工具的性能以及HLA-B * 35:01和HLA-B * 57:01等位基因的(EB vs N)方法的性能可能归因于天然加工的肽数据。
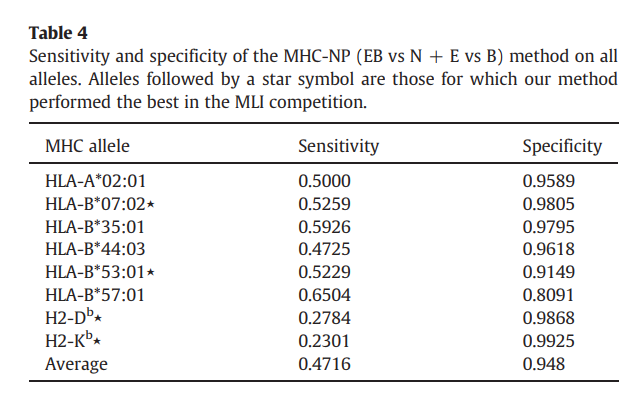
最后,表4报告了MHC-NP在所有等位基因上的敏感性和特异性。拟议的方法实现了0.4716的平均灵敏度和0.948的令人印象深刻的平均特异性。回想一下,敏感性和特异性取决于决策阈值。考虑到MHC-NP的高特异性和低敏感性,可以合理地认为可以选择MHC-NP的决策阈值以实现更好的特异性/敏感性折衷。给定表2中报告的AUC结果,显然存在这样的阈值。
五、讨论
我们提出了一种新的方法,MHC-NP,用于预测由MHC途径天然加工的肽。 我们表明,MHC-NP优于基于MHC结合亲和力的最新方法。 此外,这些结果支持以下假设:与MHC分子牢固结合的肽具有被自然加工到细胞表面的更大倾向。 在没有洗脱的肽数据的情况下,MHC结合亲和力的预测仍然是鉴定天然加工的肽的合理方法。 此外,所提出的方法适合与最新的泛特异性MHC结合工具结合使用,以提高其预测准确性。 最后,可以通过使用有关有助于MHC途径的分子的其他数据来进一步改进该方法。
参考资料
- Giguère S, Drouin A, Lacoste A, Marchand M, Corbeil J, Laviolette F. 2013. MHC-NP: predicting peptides naturally processed by the MHC. J Immunol Methods 400-401:30-6.
