【4.4.3.2】NetMHCIIpan-3.2
主要组织相容性复合物II类(MHC-II)分子在专职抗原呈递细胞的表面上表达,其中它们向T辅助细胞展示肽,其协调许多宿主免疫应答的发作和结果。因此,了解MHC-II分子将呈递哪些肽对于理解T辅助细胞的活化是重要的,并且可以用于鉴定T细胞表位(T-cell epitopes)。我们在这里提出了两种MHC-II-肽结合亲和力预测方法NetMHCII和NetMHCIIpan的更新版本。这些是使用从包含HLA-DR,HLA-DQ,HLA-DP和H-2小鼠分子的免疫表位数据库(Immune Epitope Database)获得的定量MHC-肽结合亲和力数据的扩展数据集构建的。我们表明,使用这种扩展数据集的训练改善了两种方法的肽结合预测的性能。两种方法均可在 www.cbs.dtu.dk/services/NetMHCII-2.3 和 www.cbs.dtu.dk/se rvices/NetMHCIIpan-3.2 上公开获取。
缩略语:
- AUC, area under the receiver operating characteristics curve;
- H-2, histocompatibility 2;
- HLA, human leucocyte antigen;
- IEDB, Immune Epitope Database;
- LOMO, leave-one-molecule-out;
- MHC-II, MHC class II;
- MHC-I, MHC class I;
- MHC, major histocompatibility complex;
- PFR, peptide flanking regions;
- UPGMA, unweighted pair group method with arithmetic mean
一、前言
主要组织相容性复合物II类(MHC-II)分子存在于抗原呈递细胞的表面,在那里它们将来自细胞外蛋白的肽呈递给T辅助细胞。许多肽-MHC复合物呈现在表面上抗原呈递细胞,但只有被T细胞受体识别的肽才会引发免疫反应,并被称为T细胞表位。鉴定T细胞表位对于细胞免疫和基于肽的诊断,治疗和疫苗的设计的一般理解是重要的。MHC-II分子是由a链和b链组成的异二聚体糖蛋白。在人类中,这两条链在人类白细胞抗原(HLA)基因复合物中编码,位于三个基因座之一,称为HLA-DR,-DP和-DQ。在小鼠中,MHC-II链编码为组织相容性(histocompatibility) 2(H-2)位点。每个基因座由许多不同的等位基因变体组成,这使得MHC-II分子具有高度多态性。由MHC-II分子呈递的肽结合由MHCα-和b-链残基形成的结合沟。肽结合沟的两端是开放的,因此可以使不同长度的肽结合。尽管MHC-II分子可以容纳可变长度的肽,但自然界中发现的最丰富的肽长度为13至25个残基。 主要与MHC结合沟相互作用的肽配体部分称为肽结合核心,通常为9个氨基酸长,在P1,P4,P6和P9位具有锚定残基。肽-MHC结合亲和力主要是由肽结合核心的氨基酸序列决定。然而,已经显示结合核心任一侧上的肽侧翼区(PFR)影响肽-MHC结合,从而最终也影响肽免疫原性。
因此,有许多因素使得难以预测肽结合对MHC-II分子的亲和力,包括:
- MHC-II分子的多态性,
- 肽长度的变化,
- PFR的影响
- 正确肽结合核的鉴定
所有这些因素使预测肽结合对MHC-II分子的亲和力的任务复杂化; 因此,与MHC I类(MHC-I)肽结合预测方法相比,大多数方法仍具有低性能。 早期的研究表明,NetMHCII和NetMHCIIpan的预测性能取决于肽结合数据的数量,因此,如果在扩展的肽结合数据集上进行再培训,可以预期这两种方法的性能会有所改善。 我们在这里调查是否确实如此。
由于潜在结合肽的多样性,识别T细胞表位是困难的。然而,由于肽-MHC结合是T细胞免疫原性的先决条件,多项研究表明,MHC肽结合强度和肽免疫原性之间存在很强的相关性。因此需要准确的和可靠的肽结合亲和力预测方法,可用于计算机筛选肽,目的是鉴定与给定宿主中的MHC-II分子匹配的T细胞表位。鉴于此,已经开发了许多不同的方法,包括NetMH-CII, NetMHCIIpan,TEPITOPE, TEPITOPEpan, PROPRED, RANKPEP和SVRMHC。 NetMHCII和NetMHCIIpan都被证明是预测结合对MHC-II分子的亲和力的最佳方法之一。使用NNAlign框架训练这两种方法,这些方法基于人工神经网络的集合,这些人工神经网络训练有关免疫表位数据库的定量肽结合亲和力数据( IEDB)。 NetMHCII和NetMHCIIpan之间的主要区别之一是NetMHCII是每个MHC分子的单个网络的集合,而NetMHCIIpan包含单个通用网络,可以预测已知蛋白质序列的所有MHC分子的肽结合亲和力。
NetMHCII和NetMHCIIpan预测肽结合对MHC-II分子的亲和力,包括HLA-DR,HLA-DQ,HLA-DP和H-2小鼠分子。 两种方法之间的主要区别在于NetMHCII仅预测与其已经训练的MHC分子的肽结合亲和力,而NetMCHIIpan可以预测具有已知蛋白质序列的任何MHC分子的肽结合亲和力。 如上所述,MHC结合强度和肽免疫原性之间存在很强的相关性,这两种方法已被广泛用作鉴定T细胞表位的指南,可用于设计基于肽的诊断,治疗和疫苗。。
在本文中,我们提出了我们的绑定亲和力预测方法NetMHCII和NetMHCIIpan的更新版本,这些方法是在IEDB上进行的> 100 000个定量多肽结合测量的扩展数据集上进行了训练,包括36个HLA-DR,27个HLA-DQ, 9个HLA-DP,以及8个小鼠MHC-II分子。 然后,我们使用一组大规模基准来评估这些新版本的性能,以研究扩展数据集如何提高两种方法的预测性能。
二、材料和方法
2.1 数据集
用于生成NetMHCII和NetMHCIIpan新版本的数据集包含2016年从IEDB(www.iedb.org)检索到的肽-MHC II结合亲和力。所有数据点都是实验性IC50结合值,其被对数转换为对数关系 1 -log(IC50 nM)/ log(50 000),在0和1之间的范围内,如Nielsen等人所述。 2016年数据集包含134 281个数据点,涵盖36个HLA-DR,27个HLA-DQ,9个HLA-DP和8个H-2分子。如Nielsen等人所述,通过聚类肽的共同基序将数据集分成五组,并将这五组用于五重交叉验证。 2016年数据集可在 www.cbs.d tu.dk/suppl/immunology/NetMHCIIpan-3.2 上公布。用于开发NetMHCII和NetMHCIIpan之前版本的数据集可在 www.cbs.dtu.dk/suppl/immunology/NetMHCIIpan-3.0 上获得。
表1中显示了2013年和2016年数据集中包含的数据摘要,补充材料中提供了2016年完整数据集的说明(表S1)。

2.2 网络训练 Network training
NetMHCII方法按照Nielsen和Lund的描述实施,NetMHCIIpan方法按Andreatta等人的描述实施。 NetMHCII是一种等位基因特异性方法,包含数据集中每个MHC分子的特定预测因子,它仅预测在训练数据中发现的MHC分子的结合亲和力,而NetMHCIIpan是泛特异性方法,其可以预测具有已知蛋白质序列的任何MHC分子。为了实现其泛特异性,NetMHCIIpan结合了有关MHC-II分子的信息,使用的伪序列(pseudo sequence)包含被认为对肽结合很重要的残基。该假序列使用Karosiene等人描述的方法构建,并且由34个残基组成:来自α-链的15个残基和来自b链的19个残基。两种方法都使用五重交叉验证设置进行训练。对于每个折叠,我们生成一个单独网络的网络集合,使用10种不同的初始配置,在使用10,15,40和60个隐藏神经元的500个周期的训练,产生总共40个网络。这是针对五个训练/测试集组合中的每一个进行的,导致总共200个网络。使用BLOSUM50矩阵编码肽和MHC假序列,并使用在结合核心两端的三个氨基酸的最大窗口上的平均BLOSUM分数编码PFR (原来是这样,采用的平均值,所以3个变成了一个输入)。对于每个肽核心,神经网络由肽核心(9 * 20 = 180个输入),PFR(2 * 20 = 40个输入),肽长度(2个输入),C末端长度和N末端PFR组成( 2 * 2 = 4个输入),导致NetMHCII的总输入值为226,NetMHCIIpan的输入值为906(来自伪序列的额外34 * 20 = 680输入值)。
2.3 绑定核心预测 Binding core predictions
为了改进绑定核心预测,我们在NetMHCII和NetMHCII-pan中都包括偏移校正步骤。 我们遵循Andreatta等人描述的程序,我们使用肽-MHC-II复合物的51个晶体结构的基准数据集评估了这种偏移校正的性能。
2.4 绩效衡量标准 Performance measures
使用接收器操作特性曲线下面积(area under the receiver operating characteristics curve, AUC)测量不同方法的预测性能。 为了将肽分类为结合物和非结合物,使用500nM的结合阈值,将所有肽的IC50结合值<500nM分类为结合物。 本文中显示的所有性能值是仅使用具有超过20种肽和至少4种结合物的MHC分子的AUC性能的平均值。
2.5 丢一分子网络训练 Leave-one-molecule-out network training
为了评估NetMHCIIpan在分子不是训练数据的一部分的情况下的预测性能,应用了leave-one-molecule-out(LOMO)方法。 为了估计MHC分子X的LOMO性能,使用上面的五重交叉验证设置训练NetMHCIIpan网络。 在LOMO交叉验证设置中,从训练集中删除了来自分子X的所有结合数据,并且所有测试集仅包括来自分子X的结合数据。该设置确保该方法在没有肽与分子结合的情况下进行训练。 因此,它可用于评估该方法预测未表征的MHC-II分子的肽结合的能力。
2.6 最近邻距离计算 Nearest neighbour distance calculation
使用关系
$$ d = (s(A,B))/ (\sqrt{s(A,A) * s(B,B) }$$
从HLA假序列的比对得分估计最近邻距离。 在该方程中,(A,B)分别是MHC分子A和B的假序列之间的BLOSUM50比对评分。
从具有至少50个数据点和至少10个结合物的分子子集中找到最近邻居。
2.7 序列标识
使用200,000个天然随机15-mer肽,顶部1%最强预测结合物的预测结合核构建序列标志,并使用具有默认设置的SEQ2LOGO显现。
2.8 生成HLA-II距离树
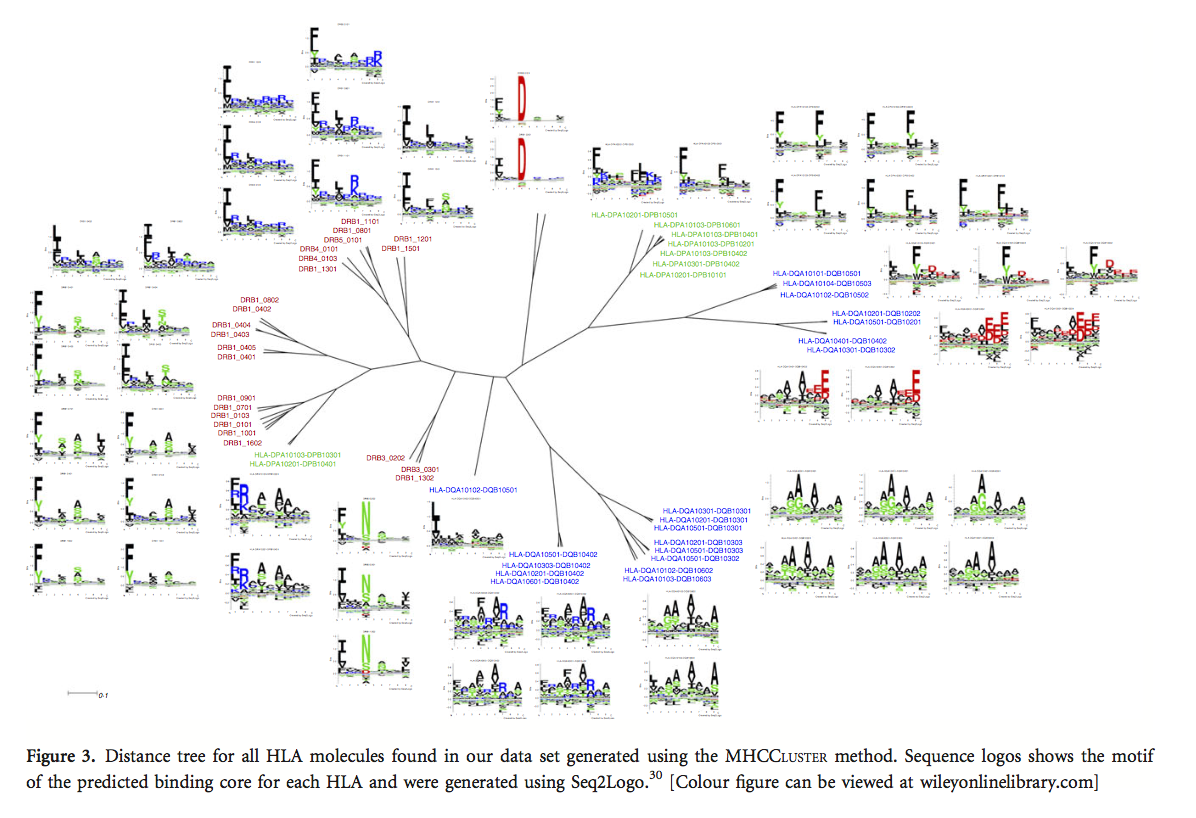
使用MHCCLUSTER为我们的数据集中的每个HLA-DR,-DQ和-DP分子生成HLA-II距离树。为了构建树,我们使用新版NetMHCIIpan,预测了200 000天然随机15-mer肽的结合亲和力。 。 然后我们使用MHCCLUSTER来发现任何两个MHC分子之间的功能相似性。 MHCCLUSTER通过校正预测的最高10%最强结合肽的结合,来计算两种MHC分子之间的相似性。 使用MHCCLUSTER中的bootstrap方法,我们生成了100个距离矩阵,并使用带有算术平均聚类的未加权对组方法将它们转换为距离树。 然后将这些树组合成一致树(consensus tree),并在SPLITSTREE中显示。如上所述构建序列标识。
2.9 T细胞表位基准
从IEDB下载通过多聚体/四聚体染色测定法鉴定的一组MHC-II限制性T细胞表位。仅包括完全类型的限制;也就是说,HLA-DQ和HLA-DP的完全键入的a-和b-链,以及HLA-DR的完全类型的b-链(其中a链是不变的)。排除了含有非天然氨基酸的表位。此外,排除了与训练数据中的肽相匹配的表位。通过将注释的IEDB蛋白ID与NCBI蛋白数据库作图来鉴定每个表位的来源蛋白序列。最终的验证数据集由1698个表位组成,限于33个不同的MHC-II分子。为了进行性能评估,将表位来源蛋白分成外表面长度的重叠肽,计算每个表位-MHC对的AUC和Frank值,将表位注释为阳性,将所有其他表示为阴性。这里,Frank是具有高于阳性肽的预测分数的肽的数量,与源蛋白质中包含的肽的数量的比率。因此,如果阳性肽具有源蛋白中所有肽的最高预测值,则Frank值为0,并且在相同数量的肽与阳性肽相比具有更高和更低预测值的情况下,值为0 5 。
三、结果
3.1 在共享评估集上比较NetMHCII和NetMHCIIpan
使用2016年的数据集,我们使用五重交叉验证设置对NetMHCII15和NetMHCIIpan11进行了再训练,以生成这些方法的两个新版本,名为NetMHCII-2.3和NetMHCIIpan-3.2。 然后,我们研究了这些新版本与先前版本(NetMHCII-2.2和NetMHCIIpan-3.1)相比如何在2013年数据集上进行了培训。 为了进行比较,我们使用了相同的五重交叉验证设置,并比较了2013年和2016年数据集之间常见的肽数据点。 该分析的结果如表2所示。
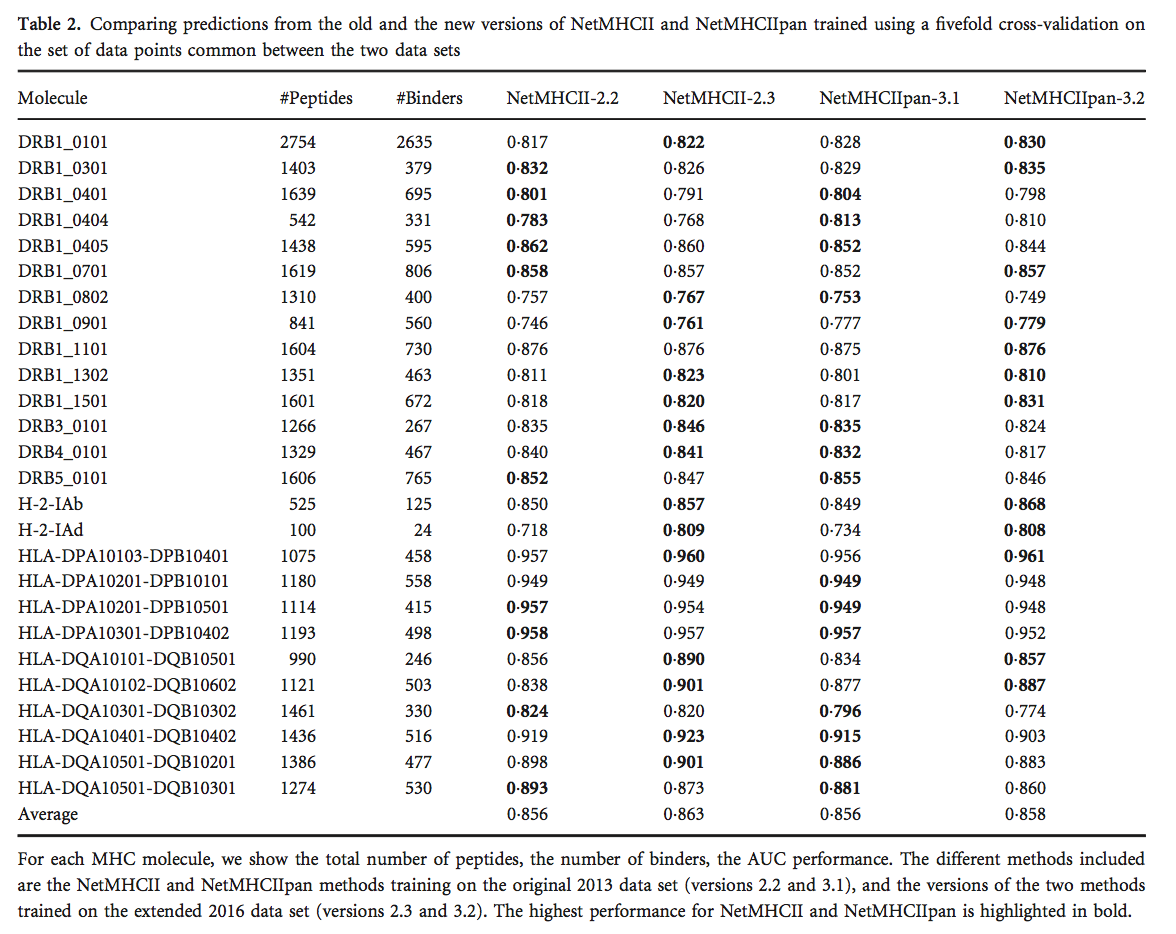
与旧版本相比,NetMHCII和NetMHCIIpan的新版本提高了性能(表2); 但是性能增益没有统计学意义(两种情况下P> 0 1)。 另一个有趣的观点是等位基因特异性NetMHCII-2.3获得了比泛特异性NetMHCIIpan-3.2更高的平均性能,但这种效应将在后面讨论。
3.2 NetMHCIIpan对常见MHC分子新数据点的表现
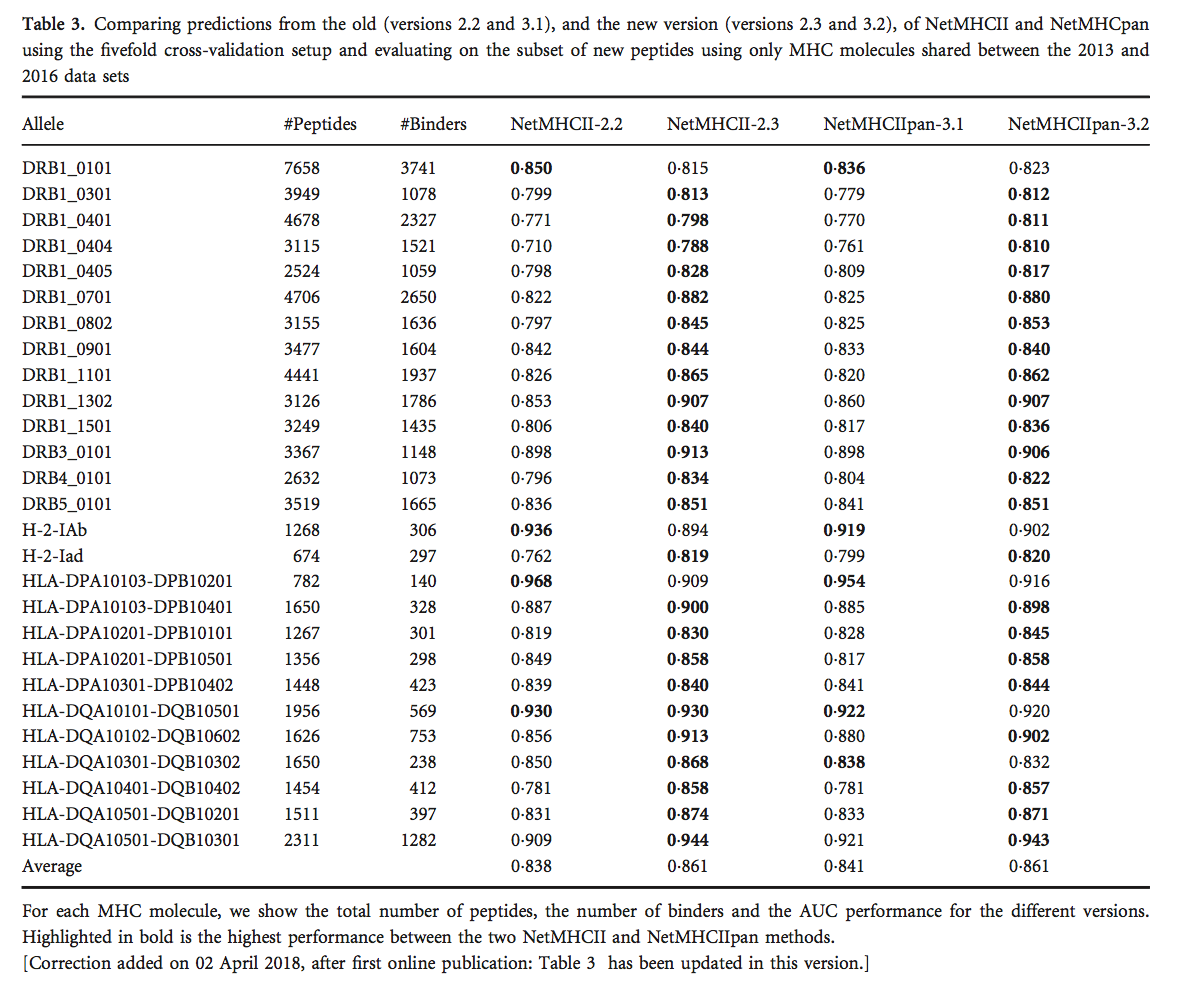
使用五重交叉验证设置,我们然后仅使用新肽的子集来评估两个版本的NetMHCII和NetMHCIIpan的性能,这些新肽用于新旧数据集之间常见的MHC分子。 该分析的结果如表3所示,它表明新版本的预测性能有显着提高(NetMHCII,P <0 001和NetMHCIIpan,P <0 0003,使用配对t检验)。 该结果强调了扩展训练数据大小的重要性,即使对于先前表征的MHC分子也是如此。 [更正于2018年4月2日增加,首次在线发表后:在前一句中,P <0 005和P <0 001分别被修正为P <0 001和P <0 0003。
3.3 绑定核心预测
我们评估了Andreatta等人描述的两种更新的MHC-II结合预测方法对肽-MHC晶体结构数据集的结合核心鉴定的准确性。总体而言,我们发现(i)包含偏移校正具有 对两种方法的结合核心鉴定的准确性产生实质性影响,以及(ii)与早期版本相比,两种方法的总体准确性得到改善。 有关详细信息,请参阅补充材料(表S2)。
3.4 执行共识方法 Performance of a consensus method
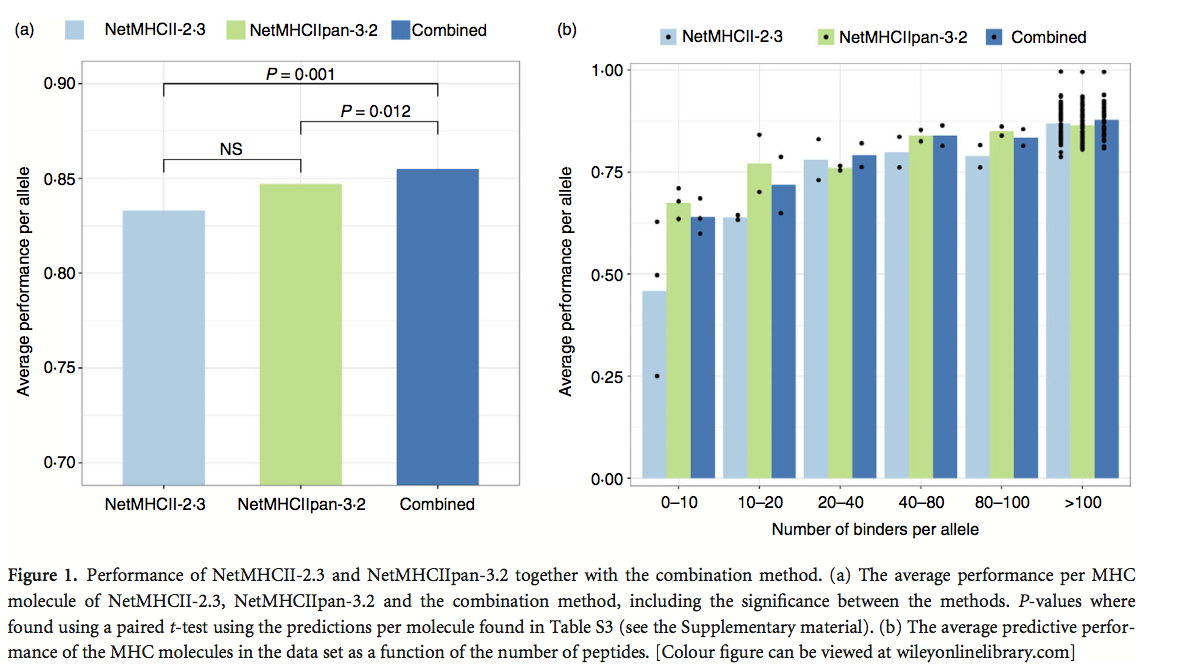
为了预测对MHC-I的结合亲和力,已经表明NetMHC和NetMHCpan的预测的简单组合比单独使用每种方法提供了更高的性能。因此我们对NetMHCII-2.3和NetMHCIIpan-3.2的预测进行了类似的组合,调查是否可以使用这种共识方法改善MHC-II的表现。在共识方法中,我们使用来自NetMHCII-2.3和NetMHCIIpan-3.2的预测分数(0和1之间的值)的平均值来定义共识方法。该分析的结果如图1所示,详细的性能值见补充材料(表S3)。图1(a)显示NetMHCII-2.3和NetMCHII-pan-3.2的组合与每种方法相比具有显着改善的性能,图1(b)显示NetMHCIIpan-3.2优于NetMHCII-2.3,尤其在数据集中仅发现少量肽的MHC分子。
3.5 NetMHCIIpan对以前无特征的MHC分子的表现
对于NetMHCIIpan,我们还测试了不属于2013年数据集的MHC分子的性能(见表4)。 正如预期的那样,我们观察到,与先前版本的NetMHCIIpan相比,NetMHCIIpan的新版本的预测性能显着提高(P = 3.6 * 10^5,使用配对t检验); 因此,该结果证明了扩展训练数据的同种异型覆盖范围的重要性。

3.6 留下一分子的表现 Leave-one-molecule-out performance
泛特异性方法能够对未表征的MHC分子进行预测,因此为了评估NetMHCIIpan方法在这些情况下的预测性能,我们进行了LOMO实验。 在LOMO中,MHC分子的结合数据被排除在训练之外,然后仅使用所讨论的MHC分子的结合数据评估所得模型(详情参见材料和方法)。 LOMO实验是针对2013年和2016年数据集之间共享的所有MHC分子进行的,并且对两个数据集之间共享的肽进行了性能评估。 表5中显示了该LOMO基准的结果,以及MHC分子与根据材料和方法中描述的,最近邻序列相似性估计的两个训练数据集中的每一个的伪距离。

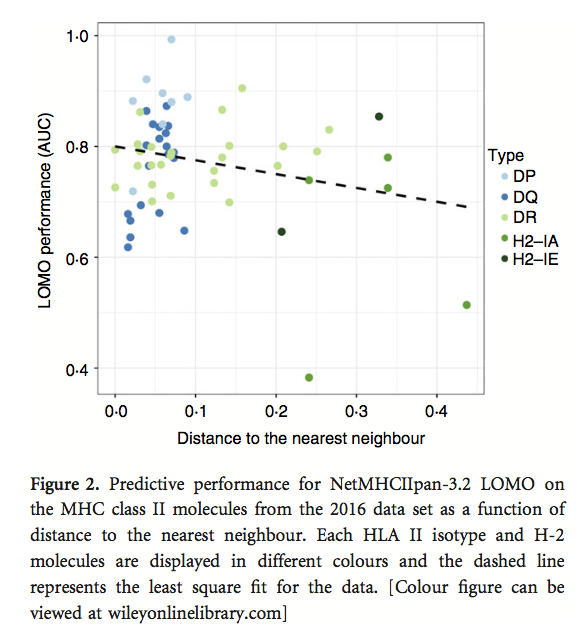
表5显示与netMHCIIpan-3.1-LOMO相比,NetMHCII-pan-3.2-LOMO的性能提高。 对于伪序列(pseudo sequenc)距离减少的MHC分子,这种增益通常最明显。
为了进一步研究最后一次观察,LOMO性能评估扩展到包括2016年数据集中的所有MHC分子。 该分析的结果如图2所示,其中散点图显示了训练数据集中距离最近邻居的距离与LOMO性能之间的关系。 用于创建图2的完整数据可以在表S4中找到。 该图显示HLA-DQ和HLA-DP分子具有接近的最近邻居,而HLA-DR和H-2分子倾向于具有更远的邻居。 该图还显示了LOMO性能与训练数据中距离最近邻居的距离之间的相关性较弱但具有统计学意义(P = 0.04,具有精确置换测试)。 这与MHC-I和MHC-II分子的早期发现一致,并显示泛特定方法的预测性能如何取决于与最近邻居的距离。
3.7 HLA分子的距离树
在达到NetMHCIIpan的最终重新训练版本之后,我们接下来使用MHCCLUSTER方法来评估2016年训练数据中包含的HLA分子之间的结合基序的相似性。简而言之,MHCCLUSTER方法使用大量随机天然肽的预测结合值之间的相关性来估计两个MHC分子之间的相似性。如果两个分子具有完美的结合特异性重叠,则相似性为1;如果两个分子没有特异性重叠,则相似性为1(详情参见材料和方法)。比较2016年训练数据中任何两个HLA II类分子之间的结合模式相似性,我们构建了图3中所示的距离树。该图证实了Karosiene等人的早期发现:11(i)不同的基因座显示有限结合偏好重叠,(ii)与HLA-DQ和HLA-DR相比,HLA-DP的多样性较少,(iii)HLA-DQ的多样性可大致分为三组;优选带有朝向C-末端的带负电荷的氨基酸,一个优先于C-末端带正电荷的氨基酸,另一个优先选择锚定位置的小氨基酸。
3.8 T-cell epitope benchmark
我们接下来评估了两种NetMHCIIpan方法对IEDB T细胞表位数据集的预测性能。我们向IEDB询问了通过四聚体/多聚体染色鉴定的MHC-II限制性表位,其是具有已知MHC限制的表位鉴定的参考标准。对于每个表位-MHC-II对,我们通过预测所有重叠肽的表位的MHC-II限制性元件的结合亲和力来计算两种NetMHCIIpan方法的AUC和Frank值,其长度与表位相同。源蛋白质序列,将表位注释为阳性,剩余的肽注释为阴性。该注释非常严格,因为共享相同配体结合核的肽被计为阴性,即使它们可以由人MHC分子呈递;因此,设置很可能会低估预测性能。该分析的细节见表S5,结果总结在图4中。
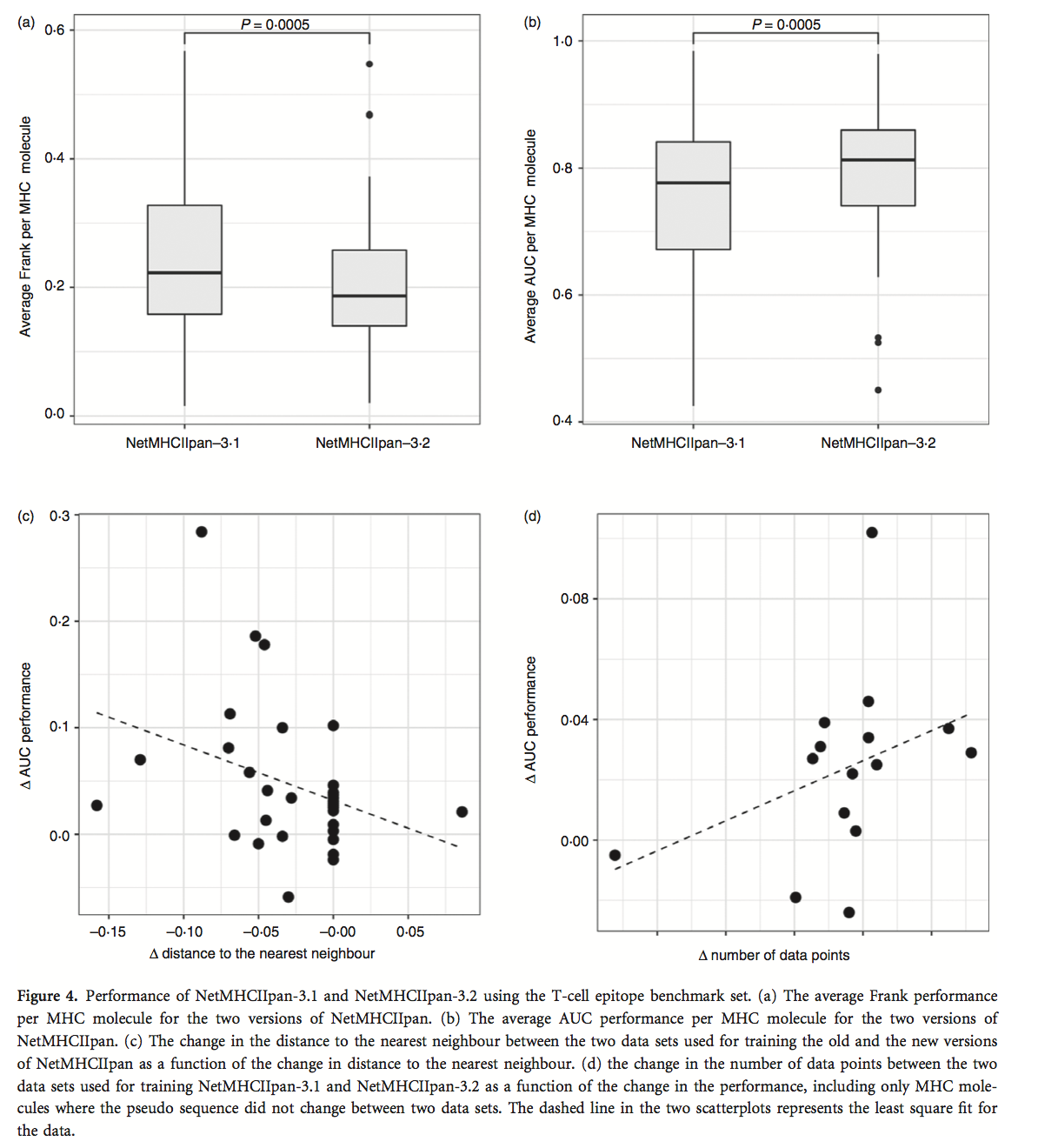
如果阳性肽具有源蛋白中所有肽的最高预测值,则Frank值为0,并且在相同数量的肽与阳性肽相比具有更高和更低预测值的情况下,值为0。图4(a)显示NetMHCIIpan-3.1的Frank得分明显低于NetMHCIIpan-3.1。它进一步表明,NetMHCIIpan-3.2具有<0.2的中值,表明如果根据其预测的肽结合亲和力进行分选,则在源蛋白的前20%肽中发现阳性肽。图4(b)显示与NetMH-CIIpan-3.1相比,NetMHCIIpan-3.2的AUC性能显着改善。我们推测,NetMHCIIpan-3.2预测性能的增加可归因于至少两个因素,即在训练数据中包含额外MHC-II分子的结合数据,以及MHC-II数据点数量的扩展已经包含在旧训练数据中的分子。图4(c,d)量化了这两个因素确实有助于提高性能。图4(c)显示了作为查询分子与训练数据最近邻居的距离变化的函数的性能增益。从该图中,我们看到预测性能的增益与最近邻距离的减小有关,因此与在新数据集中包含额外MHC-II分子的结合数据直接相关。图4(d)显示了作为用于训练的两个数据集之间的数据点数量变化的函数的性能增益。我们这里仅包括用于训练NetMHCII-pan-3.1和NetMHCIIpan-3.2的两个数据集之间共享的分子,因为我们在之前的分析中证明了到最近邻居的距离如何影响性能。图4(d)显示,性能增益与给定MHC分子数据点数的变化相关。这表明新NetMHCIIpan版本的性能增益也受到2013年数据集中已包含的分子数据点数量增加的推动。图4(c,d)中具有增加的最近邻距离和减少的数据点数的一个数据点对应于在2016数据集中去除了错误数据的HLA-DPA10103-DPB10201分子。
四、讨论
编码MHC-II分子的基因组区域极其多态,包含数千个等位基因,因此很难产生足够的实验数据来表征所有现有MHC-II分子的肽结合偏好。 因此,大多数MHC-II分子仍仅用极少数或无结合数据表示,限制了先前结合亲和力预测方法的覆盖范围和性能。 因此,我们使用更新和扩展的数据集更新了我们的两个绑定亲和力预测方法NetMHCII和NetMHCII-pan。 对于几个大规模基准测试,这改善了两种方法的预测性能。
4.1 比较NetMHCII和NetMHCIIpan
使用旧数据集和更新数据集共享的数据点,我们首先比较了NetMH-CII和NetMHCIIpan的不同版本。我们展示了这些方法的新版本如何优于NetMHCII和NetMHCIIpan的先前版本。然后,我们仅使用“新”肽评估两种方法的性能,对于旧的和更新的数据集涵盖的MHC分子。该分析的结果表明,该数据集上的两种方法均获得了预测性能的显着改善,支持扩大训练数据大小的重要性,即使对于已经以结合数据为特征的MHC分子也是如此。在评估新肽时,必须记住,MHC结合预测因子通常用于选择肽进行实验验证,而新数据集可能不如在给定集合的整个空间内采样生成的历史数据集多样化蛋白质序列
NetMHCII和NetMH-CIIpan之间的主要区别在于NetMHCII是针对每个MHC分子单独训练的等位基因特异性方法,而NetMHCIIpan是泛特异性方法,其包含使用来自数据集中所有MHC分子的信息的单个网络集合。 。因此,我们期望等位基因特异性方法优于MHC分子的泛特异性方法,其中有足够的数据可用于准确表征结合基序,并且我们希望泛数特异性方法在数据稀少时优于等位基因特异性方法。 这正是我们在比较NetMHCII-2.3和NetMHCpan-3.2的预测性能时所观察到的。早期的工作已经显示出类似的结果,即当等位基因特异性神经网络预测算法依赖足够数量的肽结合物来实现高预测性能时。这说明了等位基因特异性方法如何仅在大量数据可用于所讨论的MHC分子,但突出了泛特异性方法的强度,这些方法可以从相关MHC分子的数据中受益,从而对数据有限的MHC分子进行可靠的预测。由于等位基因特异性和泛特异性方法之间存在差异,我们实施了两种方法的简单组合,因为这已被证明可以提高MHC-I分子的预测性能。该分析显示NetMHCIIpan-3.2优于用于MHC分子的NetMHCII-2.3,其已经用很少的肽训练,但是来自两种MHC-II方法的预测的组合仍然优于每种单独的方法。
4.2 为NetMHCIIpan剥离一分子性能 Leave-one-molecule-out performance for NetMHCIIpan
NetMHCIIpan方法的主要优势之一是它可以预测未表征的MHC分子的结合亲和力。为了评估该方法在这项任务中的表现,我们构建了一个LOMO实验,我们测试了NetMHCII-pan方法的性能,用于预测未包括在该方法的训练数据中的MHC分子的结合亲和力。从该分析中,我们可以证明泛特异性方法能够预测MHC分子的结合亲和力,其中没有可用的结合亲和力数据,并且进一步证明预测性能取决于到最近邻居的距离。最后的观察表明,通过将更多未表征的MHC分子包括在训练数据中,可以进一步改善NetMHCIIpan方法的预测性能,因此,对于尚未表征的MHC分子,以目标方式产生实验肽结合亲和力数据点是重要的。
4.3 HLA II类分子的距离树
为了理解不同组的HLA II类分子,我们使用NetMHCIIpan-3.2生成了一个虚构的距离树。 该距离树中显示的组可用于理解肽如何与不同的MHC分子相互作用,并可用于区分结合物和非结合物。 距离树也可用于鉴定具有与基于表位的疫苗的设计相关的相似特性的T细胞表位。 可以在树上观察到的另一个方面是大多数MHC分子在P1,P4,P6和P9处具有强锚定位,这在先前的研究中也已观察到。
准确预测肽结合对MHC分子的亲和力,对于理解细胞介导的免疫应答和产生更好的筛选方法,以便经济有效地鉴定免疫原性肽是重要的。因此,我们想要测试两个版本的NetMHCII-pan对T细胞表位数据集的预测性能,并且这样做我们证明了新版NetMHCIIpan与早期版本相比如何获得显着改善的预测性能。两个主要因素解释了这种性能增益:(i)包括新MHC-II分子的数据减少了到最近邻居的距离,(ii)包括增加的数据点数使得该方法能够更好地表征给定MHC的特异性-II分子。
总之,我们认为NetMHCII和NetMHCIIpan可用于改善MHC-II结合预测并降低在基于表位的疫苗设计领域工作的免疫学家的实验成本,并提高我们对pep-MHC相互作用的认识,是细胞免疫反应中的关键事件。
参考资料
Jensen, K. K., Andreatta, M., Marcatili, P., Buus, S., Greenbaum, J. A., Yan, Z., … Nielsen, M. (2018). Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology, 154(3), 394–406. https://doi.org/10.1111/imm.12889
