【6.7.2】NGS用于慢性淋巴细胞白血病(CLL)体细胞高突变(SHM)状态的评估
体细胞高突变(SHM)状态为慢性淋巴细胞白血病(CLL)(一种非常常见的成熟B细胞白血病类型)提供了重要的预后指标。由于与未突变的免疫球蛋白重链变量(IGHV)状态相关的不良预后,SHM检测作为CLL的标准治疗方法。常规上,在PCR扩增CLL细胞中的克隆免疫球蛋白重链基因重排后,使用劳动密集型且主要是模拟Sanger测序方法进行SHM测试。相比之下,下一代测序(NGS)的最新可用性允许在肿瘤性B细胞人群中进行更广泛的检测和直接鉴定克隆性免疫球蛋白基因重排。能够在基线(诊断)和治疗后设置中识别特定的克隆IGHV标签的能力,使得NGS具有独特的临床应用,例如确定SHM状况,监测最小残留疾病(MRD,minimal residual disease),克隆异质性和B细胞受体IG刻板印象(stereotypy)。我们提供了有关使用NGS确定SHM的当前实践和建议的回顾,包括困难案例的例子。
一、前言
1.1 CLL的IGHV重排和体细胞突变
IGH区基因编码免疫球蛋白(IG)重链蛋白的抗原结合可变区(V)和同型特异性(isotype-specific)恒定区(C),它们的组合产生巨大的序列多样性,这对于有效鉴定多种抗原非常重要(Tonegawa,1983年)。免疫球蛋白重链由多个基因编码,包括可变(V),多样性(D)和连接(J)基因,它们进一步组织为四个相对保守的框架区(FR1,FR2,FR3,FR4)和三个可变互补决定区(CDR1,CDR2,CDR3)(图1)。 FR1 / CDR1,FR2 / CDR2和FR3区由IGHV基因编码,而CDR3由IGHV基因的3’部分,IGHD基因和IGHJ基因的5’部分组合而成。此外,在V-D和D-J基因连接处添加了可变数量的“非模板”核苷酸(N,“non-templated” nucleotides)和回文(P,palindromic)核苷酸,从而大大增加了序列多样性。这样,对于任何给定的B细胞,CDR3序列代表重排的IGH基因的高度独特的区域。
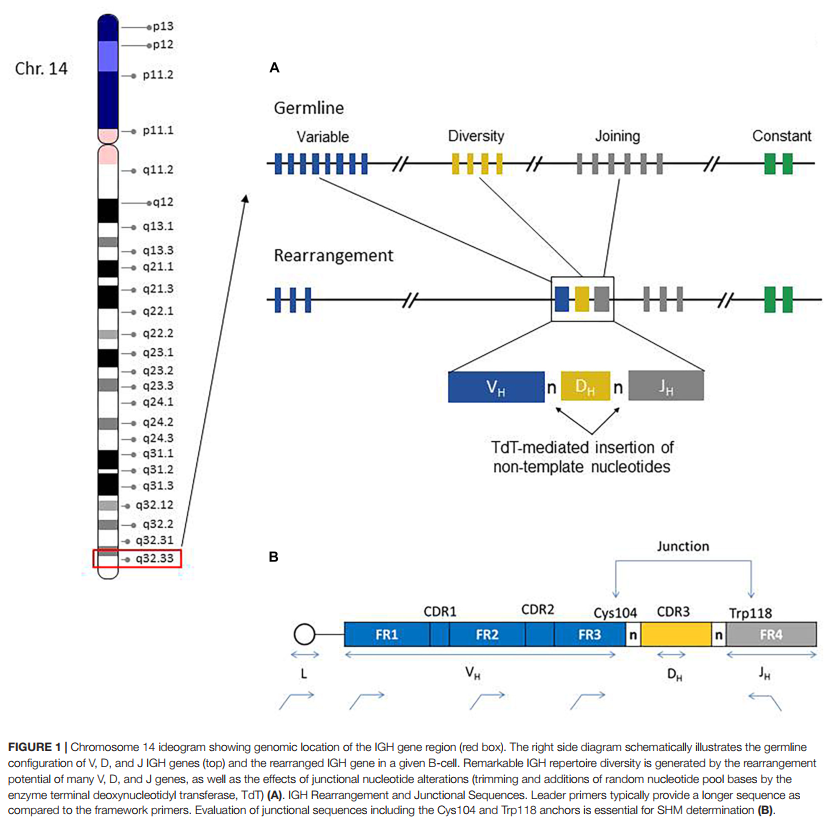
在B细胞发育期间,IGHV(n = 38-46),IGHD(n = 23)和IGHJ(n = 6)基因发生随机重排(图1)。只有那些具有生产力并因此具有功能的IG基因重排才保留在B细胞中,并与具有类似功能的重排的免疫球蛋白轻链基因产物(κ或lambda)结合形成完整的免疫球蛋白受体。没有产生IGH重排的B细胞会发生凋亡。
B细胞中IGH基因重排在暴露于抗原后会发生其他体细胞突变,这是淋巴组织生发中心反应的一部分。这些获得性或体细胞超突变(SHM)由酶激活诱导的胞苷脱氨酶(AICD)介导,并且主要涉及CDR区中核苷酸碱基的变化。 SHM发生在抗原激活的生发中心B细胞中,并通过亲和力成熟过程提高了参与多克隆免疫反应的细胞的适应性,从而优化了免疫球蛋白对抗原表位的结合(Neuberger和Milstein,1995; Di Noia和Neuberger,2007; Neuberger和Neuberger,2007)。 Pilzecker和Jacobs,2019年)。因此,SHM是体液免疫的重要方面。
作为SHM的结果,受影响的B细胞中IGH基因重排的核苷酸序列与其对应的种系对应物不同。在CLL的设置中,SHM状态用于定义不同的预后亚组。 “未突变”的克隆IGH基因重排定义为与其最接近的种系IGHV基因具有非常高的核苷酸序列同一性。相比之下,“突变” CLL的特征在于IGHV序列相对于其种系参考有一定百分比的偏差。按照惯例,CLL中SHM的状态是在诊断时识别的优势克隆种群上报告的,定义为与最接近的种系IGHV参考序列的偏差> 2%(突变状态)或≤2%(未突变状态)。一些研究证实(Damle等人,1999; Hamblin等人,1999;Kröber等人,2002),未突变的CLL通常与较差的预后相关,而突变的CLL显示出较弱的侵袭性病程。通过基于IGHV CDR3的受约束特征识别CLL的特定子集,进一步的子分类变得显而易见[请参阅以下有关B细胞受体(BCR)刻板印象的部分];这些子集在预后上也很重要,并且可能与SHM状态无关。例如,具有IGHV3-21重排的CLL病例(尤其是属于子集#2的那些)是一个例外,因为这些患者的预后较差,而与SHM状态无关(Tobin等,2002;Thorsélius等,2006; Baliakas等人,2015年)。
1.2 使用Sanger测序进行体细胞超突变测试
按照惯例,CLL中的SHM分析是使用IGHV结构域的Sanger测序(使用DNA或RNA作为起始材料)进行的。该方法被认为是确定SHM状态的金标准方法,涉及两个步骤:一种基于PCR和毛细管电泳的方法来检测克隆性,然后进行自动荧光染料终止剂Sanger测序。如所指示的,SHM测定评估CLL IGH基因与最接近的匹配的种系序列对应物之间的序列偏离水平,以便基于如上所述的2%阈值来分配未突变或突变状态。基于Web的IG种系序列数据库,例如IMGT(国际ImMunoGeneTics信息系统1)和IgBlast2,极大地促进了这一分析过程。尽管已经使用了很多年,但是由于劳动强度,技术复杂性和可扩展性的局限性,该测定在许多临床实验室中仍然不能统一进行。尽管此技术通常在单克隆IGH重排的情况下效果很好,但重排> 1的病例(多达10%的病例可见(Langerak等人,2011))由于无法识别可靠地量化单个克隆的相对丰度而极难解释。。使用Sanger测序也可能低估了> 1重排的病例的发生率。
大规模并行测序(也称为下一代测序(NGS))的最新可用性提供了明确确定各个克隆序列及其相对比例的能力。确定每个克隆的特定序列的能力提供了对肿瘤内和肿瘤间异质性以及克隆进化的更好理解。在治疗前设置环境中确定克隆特定序列的能力可以检测出治疗后可测量的最小残留疾病(MRD,minimal residual disease)。近年来,许多小组分享了他们在CLL中使用NGS测定进行SHM检测的经验(Blachly等人,2015; McClure等人,2015; Stamatopoulos B.等人,2017)。下面我们总结了这些研究的重要发现,以更好地理解CLL中基于NGS的SHM测试。此外,我们还提供了SHM测试的临床验证和报告注意事项,包括具有指导意义的临床案例。
二、NGS用于CLL的体细胞超突变(SHM)的最新经验
McClure等人研究了将NGS用于SHM分析。 (2015年),其中使用Ior Torrent PGM平台(美国马萨诸塞州沃尔瑟姆市的ThermoFisher)将Sanger测序与实验室开发的NGS策略进行了比较。 NGS数据采用多种方法组合处理,包括板载软件(on-board software)和IMGT工具,以使读数与种系IGH参考序列比对,并进行SHM百分比计算。 NGS数据的替代可视化还使用专有的自动售货(proprietary vended)软件和Integrated Genome Viewer(IGV,Broad Institute,Cambridge,MA,美国)完成,以找到克隆种群并建立IMGT比对的共有序列。虽然这两种方法均显示出可比的准确性,但NGS的独特优势包括分批处理(效率更高),直接克隆确定,成本竞争力(运行分批处理)以及在某些情况下能够识别> 1个主要的IGH重排的能力。尽管这两种方法都需要大量的用户技能和特定软件应用程序以及数据分析的经验,但是与使用Sanger方法创建干净的序列重叠群的更为费力的方法相比,NGS数据中的读取堆积很容易生成共有克隆序列。然而,在这项研究中,一些方法学上的矛盾是显而易见的,包括使用前导特异性引物进行PCR / Sanger测序,而不是使用家族特异性FR1引物进行NGS方法。 FR1引物的使用可能会导致IGH SHM计算不准确,因为需要在V结构域内放置测序引物,从而导致分析的靶区域被截短。然而,作者指出,这基本上不会影响CLL病例的突变状态分配。尽管NGS方法固有的复杂性更高,但测试性能和分析的总体动手时间却是可比的。实际上,在大多数情况下,NGS的半自动数据分析被认为可以简化分析的解释。还注意到基于PGM的方法工作流程与测定无关,因此如果同时批处理具有不同测序目标的样品,以最大化测序芯片的利用率,则可以导致实验室工作和成本效益。
Stamatopoulos B.等。 (2017)使用LymphoTrack IGH体细胞超突变检测试剂盒(Invivoscribe,San Diego,CA,United States)对纯化的CD19 + CD5 +细胞的互补DNA(cDNA)进行了检测,在近25%的CLL患者中发现了多种生产性IGH克隆重排。由于早期的测试方法很少观察到此发现,因此在排除正常B细胞的污染并使用其他生物信息学方法后对其进行了交叉检查。多个克隆的发现可能部分是由于NGS的敏感性更高。与Sanger测序相比,使用NGS可以更可靠地扩增DNA模板混合物并进行测序。此外,当通过NGS发现具有多个重排的患者使用特定VH家族前导引物扩增IGHV时,结果可以得到证实。重要的是,可以根据IGHV NGS概况将患者分为五个亚组:multiple hypermutated (M) clones; 1 M clone; mix of M and unmutated (UM) clones; 1 UM clone (including V3-21 cases); and multiple UM clones。这些亚组的疾病结局取决于克隆的类型,这取决于无治疗和总体生存率。发现使用NGS可以改善CLL的分层及其临床管理,并有助于选择新疗法的患者。 CLL中亚克隆异质性的存在也挑战了恶性淋巴疾病中当前的“单克隆性”概念,并暗示了更高的生物学复杂性,对某些患者的治疗反应和结果可能产生影响。
Blachly等。 (2015)使用下一代RNA测序(RNA-seq),并且能够从未选择的RNA-seq读数中获得IGH转录本的完整序列。发现具有高分辨率和高动态范围的RNA-seq转录组数据的多维性质,不仅对IGHV SHM状态测定有用,而且对同时评估全局基因表达以及鉴定已知和新颖的遗传变异有用。结论是,无偏见的下一代RNA-seq可以潜在地取代当前基于Sanger测序的测定法,以准确确定IGHV序列,SHM状态和其他肿瘤遗传特征。
Wren等。 (2017)致力于淋巴增生性疾病的易位和克隆性的综合检测。他们表明,利用基于探针捕获方法的EuroClonality-NGS面板,NGS可用于对V-(D)-J基因重排进行测序。该方法可同时评估克隆性,克隆关系和进化,IGHV SHM,并可为可测量的残留疾病指定靶标。可以将更全面的NGS面板设计为还包括与淋巴细胞增生性疾病的诊断和预后相关的复发性易位和遗传改变,以及在单个测定中进行IGHV SHM分析。相对无偏的分析也可以评估免疫球蛋白基因库。对免疫库进行深度测序的目的之一是评估库对抗原环境的反应。 Six等人综述了统计和建模方法。 (2013年)分析淋巴样增生中的免疫球蛋白和T细胞库(Six等,2013)。
2.1 基于NGS的IGHV SHM测试支持的其他应用
MRD (Monitoring Minimal residual disease )使用NGS进行CLL中评估,通常使用诊断时确定的用于SHM测定的相同克隆(Logan等,2011)。与目前公认的MRD评估方法(如等位基因特异性寡核苷酸PCR(ASO-PCR)和多参数流式细胞术(FCM))相比,基于NGS的方法为MRD分析提供了独特的优势。 ASO-PCR是高度敏感的,但鉴于艰巨的技术要求(包括患者克隆特异性引物的合成和验证),通常不进行。尽管FCM广泛可用,但免疫表型测定法通常具有有限的分析灵敏度。另一方面,基于NGS的IGH VDJ片段重排测定结合了等位基因特异性PCR和FCM测定的灵敏度和广泛的适用性,同时使用共有的PCR引物,避免了患者特异性引物的产生。 Rawstron等人还描述了使用多参数流式细胞仪和高通量测序进行高灵敏度CLL-MRD分析的组合方法。 (Rawstron et al。,2016)。
B-Cell Receptor IG Stereotypy
CLL中基于下一代测序的分析也可用于受体定型分析。 CLL中的受体定型定义为在无关的CLL患者中几乎相同的BCR免疫球蛋白(IG)肽基序的存在。鉴于IG重排的多样性很大,任何两名拥有相同或高度相似的克隆IG基因产物的CLL患者的统计概率都应该非常小,但是令人惊讶的是,根据CLP的相似性,可以将三分之一的CLL患者分组为刻板印象的亚组。 IGH CDR3重链可变区。受体定型亚组分析是对临床和预后亚组评估的进一步完善,并改进了目前的突变和未突变CLL的二进制名称。最近的研究表明,具有几乎相同的BCR IGH谱或相同的受体刻板印象的无关CLL患者在临床特征,遗传病变,表观遗传学变化,临床过程和临床结果方面彼此具有可比性。例如,最大的定型子集是子集#2(IGHV3-21 / IGLV3-21),与SHM状况无关,其表现出剪接体遗传改变,侵袭性临床病程和不良预后。因此,除了SHM分析之外,受体刻板印象可以作为定义更多不同或同质亚组的基础。此外,具有相同刻板印象的子集可能是优化治疗方法的候选者(Tobin等人,2003; Stamatopoulos等人,2007; Agathangelidis等人,2012; Stamatopoulos K.等人,2017)。
三、IGHV SHM NGS-分析注意事项
使用NGS准确评估克隆性需要相对无偏的文库制备(即基于PCR生成的或基于捕获探针的)和所有可能的VDJ重排的高通量测序。 但是,由于IGHV重排的技术和生物学差异,这些看似简单的目标难以实现。 简而言之,与基于Sanger测序的IGHV SHM分析相比,NGS评估通常是通过使用IGH家族特异性前导序列和共有JH引物PCR预混液扩增提取的DNA来进行的。 使用基于微珠的方法纯化PCR产物,将生成的文库标准化,然后合并用于测序。 以下总结了基于NGS的SHM测试的主要注意事项。
3.1 引物
根据ERIC指南(Ghia等人,2007; Rosenquist等人,2017),应使用前导引物,以便可以获得完整的IGHV区序列(图1),并因此与最接近的种系参照物具有相同的百分比可以最精确地计算基因(即,靶标/参考核苷酸的比对数×100)(Sahota等,1996; Fais等,1998)。如果前导引物无法识别显性克隆种群,则诊断实验室也可使用构架区1(FR1)引物。通常,建议在其中一种方法无法产生信息性序列的情况下,采用利用前导和FR1引物的策略。但是,在使用FR1家族引物时,需要考虑一些其他注意事项。例如,与前导引物产生的PCR模板相反,无法使用FR1引物评估完整的IGHV区,因为未对FR1引物位点上游的约60个核苷酸碱基进行测序。因此,与前导PCR结果相比,使用FR1锚定的引物的种系同一性百分比是非常接近的(Marks等,1991; Kuppers等,1993)。 FR1和前导引物结果之间的这些微小差异可能会影响最终突变%的计算。由于FR1 PCR产物产生的核苷酸碱基分母较小(即IGHV区域长度),因此可能导致对突变百分比的轻微高估,这可能在2%的常规SHM阈值附近相关;但是,通常发现这些差异对于确定SHM状况无关紧要,并且通常不会影响最终的临床结果(突变与未突变),如McClure等人所述。 (2015)。这项研究还发现了使用FR1引物进行精确IGHV基因分配的另一个潜在问题。因为FR1家族特异性引物已整合到相应VH家族的所有基因重排中,所以不会区分引物区域内发生的任何微小片段序列差异(即等位基因多态性),从而导致最接近的种系同一性的模棱两可的结果。当识别出几个紧密相关的V基因等位基因但无法进一步描述时(例如IGHV4-34 * 01或V4-34 * 04),便可以观察到这一点。实际上,在大多数情况下,这种情况不会严重影响SHM状态的准确提供,但在解释FR1 PCR结果时需要考虑。相反,SHM对B细胞IGH分析中的靶标富集提出了潜在的重大技术问题。由于低效共有引物结合,可能无法在PCR扩增步骤中识别到IGH基因中的高度突变区域。这样的克隆因此不代表进行分析。
总之,对于精心设计的测定,重要的是包括相对全面的策略,该策略采用:
- 结合至每个V或J片段不同位置的多个引物组;
- 更长的引物设计,在高度保守的位置带有锚点;
- 或使用全长cDNA引物位于V和J区域之外的引物,这些引物不太可能发生SHM(Robins,2013)。
- 重要的是为所有7个VH家族选择前导引物,而不是仅依赖FR1,这样就不会丢失任何靶标。
- 同样,如果可能的话,最好使用两个反向引物(CH和JH),而不是仅使用一个,以确保更少的假阴性PCR结果。
3.2 读取长度 Read Length
如前所述,在NGS进行的SHM分析中,重要的是对全长读数进行分析,以便分析每个序列的真实表示。该测定需要明确读取大约350个碱基对(bp)的长度,包含整个IGHV和连接区,在大多数情况下均可获得,尽管DNA质量欠佳或降解的样品可能导致无法获得最佳读取长度。使用前导引物的商业LymphoTrack测定法(Invivoscribe Inc.,圣地亚哥,加利福尼亚州)在Illumina Miseq上产生大约300 bp长的配对末端序列读数(301×2个读数+ 8个前导引物碱基= 610 bp)。平台,以便可以在单个片段中对给定重排的整个IGHV区域进行测序。还通过生物信息学评估了最终序列的生产性重排特征(即,预测的RNA转录物中没有终止密码子,符合读框的序列,以及在各个连接末端存在保守的Cys104和Trp118残基)。本质上,从引物区域到连接的Trp118的完整读框被认为是有效的。 McClure等人报告使用Ion Torrent PGM平台读取平均350 bp长度,使用FR1 PCR引物读取Ion 400 bp试剂盒(McClure等人,2015)。对于SHM分析来说,非常短的读数分析是不可取的,并且长度小于150 bp或没有CDR3序列的序列被认为不足以进行评估。
3.3 测序平台
NGS平台的选择基于各种因素,例如所需的周转时间,样品的性质和数量,要测试的遗传变异的复杂性,实验室中预先存在的平台以及在临床实验室中实现相对简单的工作流程的能力,仅举几例。支持较长读取并具有最低可能错误率的测序平台将是首选,因为它们将提供对SHM的准确评估。在某些基因组环境中错误率较高且读取长度较短的平台很可能会产生SHM%的错误升高。具有更高的AQ得分临界值(AQ30或更高)的测序质量平台可能会提供更准确的测序数据。真正和可再现的突变率超过背景测序错误率需要仔细验证。当前,可以使用“短读”方法获得更长读长的最受欢迎的平台包括Illumina MiSeq和Ion Torrent仪器。通常,Illumina测序仪可以实现足够的双向读取(2×300 bp),并且与Ion Torrent相比,具有更低的序列错误率,但是对于适当的验证,两者都是可以接受的。长读技术方法的出现(例如Pacific Biosciences,Oxford Nanopore)可能提供了询问IGH重排全长的替代方法,但是这些系统当前不适用于临床用途或原始测序错误率不可接受,这是不理想的。
3.4 控制项 Controls
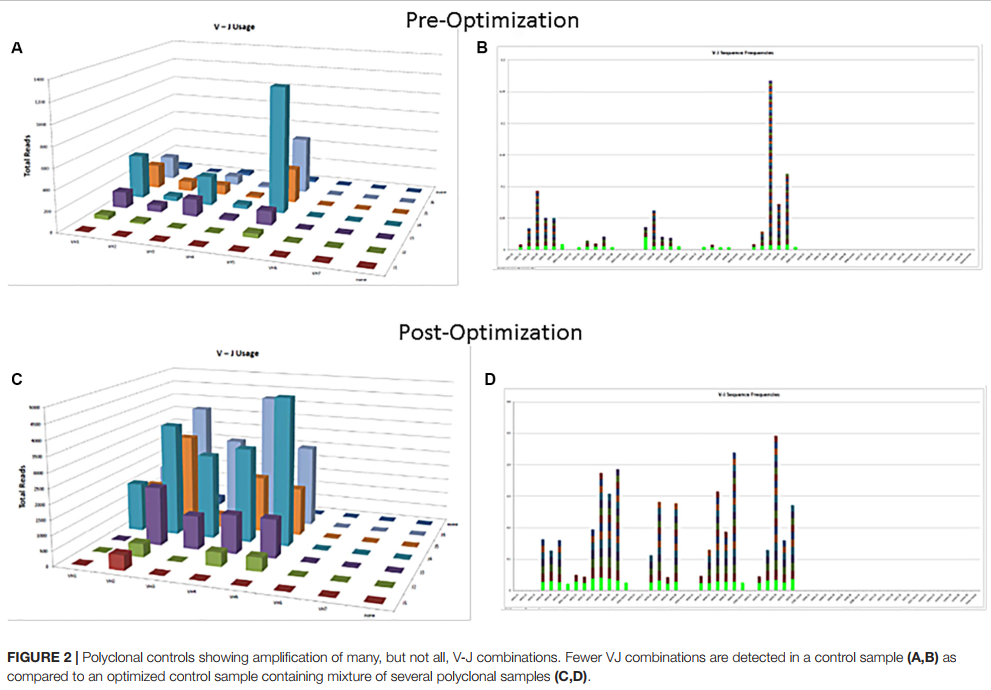
分析试剂盒(如Lymphotrack)随附的市售阳性(突变单克隆)和阴性(多克隆)对照应始终与每批一起运行。多克隆对照将有助于检查测定中使用的所有引物的性能。如果没有在市场上提供的多克隆对照中代表所有家族,则可以合并内部多克隆样品并验证其使用效果,以具有更好的V区家族代表(图2)。可以使用以前使用金标准Sanger测序分析过的已知超突变状态的已归档CLL样品,对SHM的NGS测定进行初步验证。应选择能够代表广泛肿瘤含量和样品类型[外周血(PB),骨髓(BM)等]和样品量的对照。也可以合并具有具有不同VDJ重组序列的克隆的阳性对照,以用作各种引物的对照,也可以准备一致地返回指定的克隆阈值水平(例如,总读数的10%)。家庭代表的文件对于确保底引物能很重要。
3.5 特定于样品和分析物的问题
样品类型: 新鲜[例如细针抽吸物(FNA),PB,BM,新鲜冷冻]与固定[福尔马林固定石蜡包埋(FFPE),涂片]标本:目前,已验证了用作实验室开发测试(LDT)的商业IGHV SHM测定法仅适用于包括PB和BM在内的新鲜样本类型,并且CLL群体至少构成样本的10%。最低肿瘤标准对于避免任何假阴性结果至关重要。使用FNA或新鲜冷冻样品是可行的,但需要额外的验证。由于DNA的降解或交联,目前不建议将FFPE样品用于SHM分析。
分析物类型:DNA与RNA(cDNA) Analyte Type: DNA vs. RNA (cDNA) :
当前,基于NGS的SHM商业化检测方法在大多数临床实验室中最常被验证可用于基因组DNA(gDNA)。总共需要500 ng输入gDNA输入。 Stamatopoulos B.等。 (2017)描述了使用RNA(反转录为互补DNA,cDNA)和gDNA的结果。根据基于Sanger的SHM分析的欧洲CLL研究计划(ERIC)指南,认为基因组DNA(gDNA)和cDNA均适用于IGHV分析(Ghia等,2007)。与gDNA相比,使用RNA具有优先识别功能性重排的优势,在gDNA中,非生产性IGH基因重排也将被检测到,可能需要进行额外的分析评估。相比之下,gDNA分析在技术上更易于使用,并具有更好的标本稳定性(例如,如果样品运输时间延长了)。在一项研究中,细胞的纯化对亚克隆检测的影响最小,但cDNA的敏感性比gDNA更好,因此可以检测其他克隆(Stamatopoulos B.等人,2017)
3.6 信息学
读取对齐 Read Alignment
经常通过将所得的IGHV序列与IMGT(国际ImMunoGeneTics信息系统)/ V-QUEST(V-QUEry和标准化)数据库中最接近的共有种系序列进行比较来量化SHM的程度(参见文本“脚注1”)。另外,IMGT提供了用于识别目标序列中潜在插入/缺失事件的高级工具。可以使用IMGT开发的在线工具(Yousfi Monod等,2004; Brochet等,2008)对V结构域(使用IMGT / V-QUEST)和CDR3区域(IMGT / JunctionAnalysis)进行准确而详细的序列分析。 ; Lefranc等,2015)。 IMGT / HighV-QUEST是用于分析通过高通量NGS深度测序分析获得的大量IG序列的工具(Li等,2013; Alamyar等,2012a,b)。 IMGT / HighV-QUEST算法与IMGT / V-QUEST以及集成的IMGT / JunctionAnalysis相似。 IMGT / HighV-QUEST每批可以分析大约150,000个IGV域序列,并对大约450,000个序列进行统计分析。 IMGT / V-QUEST的输出包含11个压缩文件,这些文件具有有关V,D,J基因排列,新等位基因,详细的连接分析体细胞突变以及其他数据的信息。
或者,可以使用IG序列分析工具IgBLAST(参见文本“脚注2”)查看与种系V,D,J基因,连接细节,IGV框架和互补决定区的匹配。 IgBLAST可用于分析核苷酸和蛋白质序列。它可以批量处理序列。 IgBLAST还可以用于同时对种系和本地IGH数据库进行搜索,以减少丢失种系V基因匹配的机会(Ye等人,2013)。尽管返回相同或高度相似的结果,但每个数据库及其关联的生物信息学工具集都是不同的,有时在分析给定的IGHV序列时可能会导致轻微的不一致。用户应在使用这些参考数据库时加深对可能变化的理解,或在适当时考虑进行跨数据库分析。
Vidjil是一个用于分析高通量V(D)J免疫库序列数据的开源平台(Duez等人,2017)。该算法旨在用于基于扩增子的测序以及基于杂交捕获的测序。该Web应用程序和源代码可从 http://www.vidjil.org/ 下载。抗原受体银河(ARGalaxy3,Antigen Receptor Galaxy)是另一种基于Web的开源工具,具有分析和可视化免疫组库高通量数据的能力,包括重新排列的V(D)J基因,SHM和类别开关重组(IJspeert等人,2017 )。
Schaller等人(Schaller等人,2015)开发了一个软件框架ImmunExplorer(IMEX),基于IMGT / HighV-QUEST输出的已处理NGS数据来分析IG和T细胞受体(TCR)多样性以及克隆性的存在。考虑到引物的效率和多样性,它有助于数据的可视化以及详细的统计分析,以识别样品中独特的克隆型。 IMEX具有克隆型跟踪,多样性分析和样品比较的功能。 IMEX可从 http://bioinformatics.fh-hagenberg.at/immunexplororer/ 免费获得。
Bystry等人在 http://bat.infspire.org/arrest/interrogate/ 上免费提供了另一个基于Web的交互式应用程序“ ARResT / Interrogate”。 (Bystry et al。,2017)。该应用程序结合了全面的计算机免疫分析所需的各种功能。 ARResT / Interrogate执行四个顺序功能。这些包括输入处理,数据选择和过滤,比较计算和详细的可视化。 ARResT / Interrogate主要基于R和Shiny(Shiny是一个R软件包,可以很容易地从R直接构建交互式Web应用程序)。分析核心使用“ data.table” R包进行有效的数据处理,并且可以对从数千种克隆型和数百万次读取生成的数据保持足够的响应度。扩展的ARResT工具包的另一个有利方面是能够上传IGH序列以查询数据库(ARResT / AssignSubsets)以获取具有预后意义的常见原型BCR IG子集。
相关序列汇总 Rollup of Related Sequences
经过大规模并行测序后,输出信息将通过生物信息学管道进行分析。配对的端读取数据经过组合,解复用和适配器修整,以生成FASTA和FASTQ文件。修剪引物序列,并除去完全相似的序列(重复)。然后使用市售软件IMGT / V-QUEST或IgBLAST对每个独特序列进行比对。例如,根据不同的IGHV-J组合对Lymphotrack的输出进行分组,并将最终数据加载到MySQL数据库中。合并几乎相同的差异(≤2个碱基)的序列,以适应PCR或测序错误。然后返回前10个合并的阅读序列以进行进一步分析,但是如果需要,可以保留对所有测序数据的访问。
体细胞超突变的计算
将包括CDR3的整个IGHV序列与最接近的种系参考序列进行比较以进行计算。 分析的序列必须包括保守的Cys 104和Trp 118残基,并进行检查以确保阅读框得以保留(即有效)且未遇到终止密码子。 同一性百分比是使用IGHV-D-J重排序列中IGHV区域突变的总数与最同源的种系IGHV基因的长度之间的比率计算得出的:
IGHV Identity(%)=100−{mutations × 100/aligned IGHV regionl ength}
将病例区分为突变和未突变组的98%临界值是任意的,最初是从较早的CLL研究中选择的,以排除多态变异序列的可能性。 按照惯例选择它是为了避免需要分析每位患者中相应的天然种系序列。 尽管也提出了用于同源性测定的其他临界值(例如97%或95%),但是为了数据的一致性和报告性,建议遵循98%的临界值(Kröber等,2002; Lin等) (2002年)。 最近,还有人提出,以连续变量而不是静态的98%截止值衡量的IGHV突变的绝对百分比偏差,可以预测接受利妥昔单抗,氟达拉滨,环磷酰胺治疗的CLL患者的生存率(Jain等人,2018年) )。 因此,除了“突变”或“未突变” SHM状态的标准定义外,最好报告IGHV同源性的实际百分比。
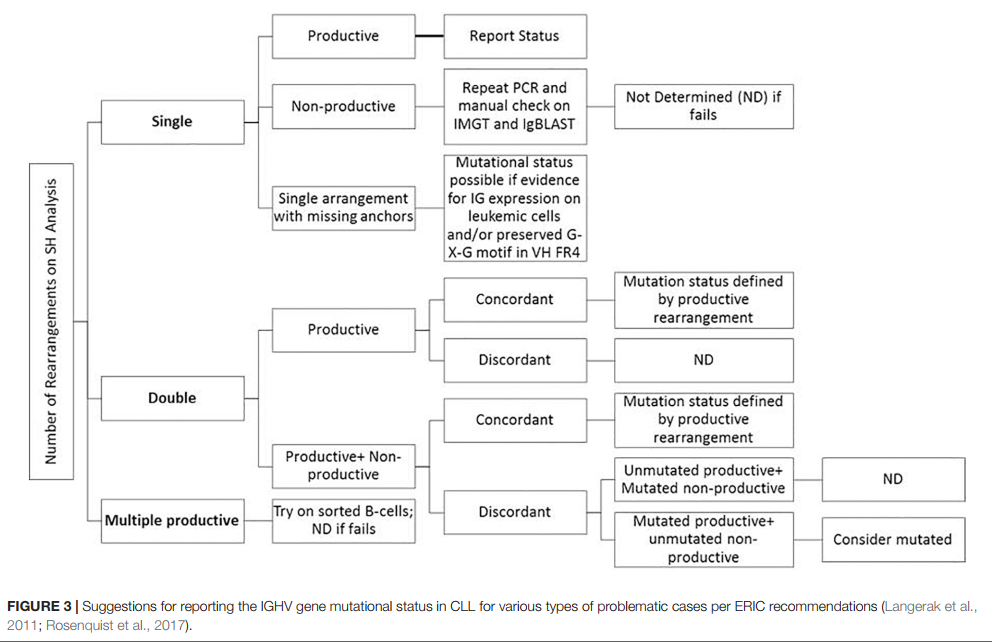
3.7 结果解释和报告
关于SHM的共识性建议最初是由ERIC(欧洲对CLL4的研究计划)于2007年提出的,随后在2011年和2017年进行了更新(Ghia等,2007; Langerak等,2011; Rosenquist等,2017)。这些指南(包括最少的报告元素)是针对Sanger测序方法而衍生的,但即使未特别提及NGS,也同样适用于基于NGS的测试。具体而言,由于NGS与Sanger测序相比具有更高的敏感性和特异性,因此可以以较高的频率看到占病例总数高达13%的“困难病例”类别。图3提供了ERIC建议的易于使用的算法表示。如果预测的RNA转录物中有一个终止密码子,该变化不是符合读框的,或者在连接处不存在保守的Cys104和Trp118氨基酸,则该序列被称为非信息性或非生产性。极短的读数(如长度少于150个碱基对的序列或没有真正的CDR3序列)也被认为不足以进行分析。在常规的临床分子诊断实践中,可以使用供应商提供的解决方案(例如LymphoTrack软件(Invivoscribe))来管理上述信息学分析因素。在此应用程序中,显示前200个最丰富的唯一序列,并将其用于计算优势克隆的总百分比,并且还提供了前10个合并序列读数的表。还指出了各种克隆型(即具有完全相同的VDJ重排但具有不同长度或突变百分比的所有唯一序列的总和)。丰度小于总读数的2.5%的克隆可以安全地作为背景正常B细胞污染而忽略不计,尤其是如果分析前已使用纯度大于98%的B细胞富集处理。 Lymphotrack软件整合了IgBLAST分析,但根据ERIC的建议,许多实验室还选择使用IMGT数据库进行分析,以计算与种系参考值相比的突变百分比。
四、案例案例
以下三个案例重点介绍了NGS在IGH SHM分析中的应用。
4.1 情况1:两次生产和协调重排 Two Productive and Concordant Rearrangements
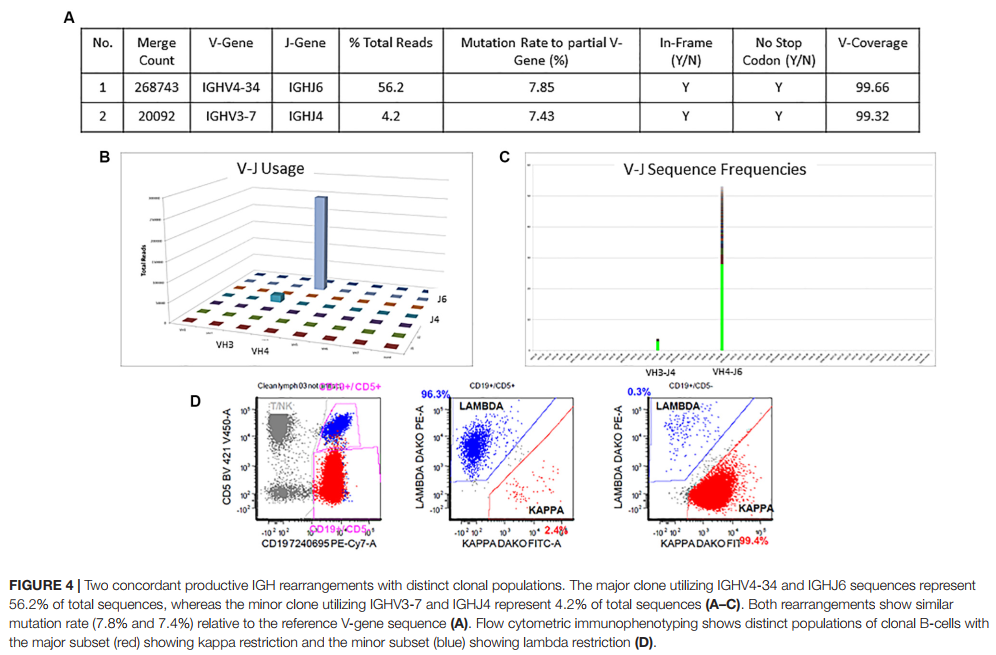
情况1的详细Lymphotrack输出可以在图4A–C中看到。总读取计数为477734,而最小可报告读取计数为180,000。在这种情况下,有两个生产性克隆,因为它们都只有框内突变,没有任何终止密码子产生。显性克隆构成总读取的56%,具有V4-34和J6的使用率和7.9%的突变率。较小的克隆-占总读取量的4%,使用V3-7和J4,突变率7.4%。两个克隆均具有生产性重排和一致的突变状态,即两个克隆均被突变。这种情况的最终解释是:IGHV状态突变。此类案例占每个ERIC的CLL案例的0.7%。有趣的是,由于它们的轻链差异,可以在流式细胞仪上分别清楚地识别这两个克隆(图4D)。显性克隆具有CD5和CD19的较暗共表达,并且受kappa链限制,而较小的克隆显示CD5和CD19的明亮共表达,并且受lambda链限制。这是一个示例,显示了流式细胞术发现与IGHV基因重排之间的相关性,可以更好地了解疾病过程及其克隆结构。
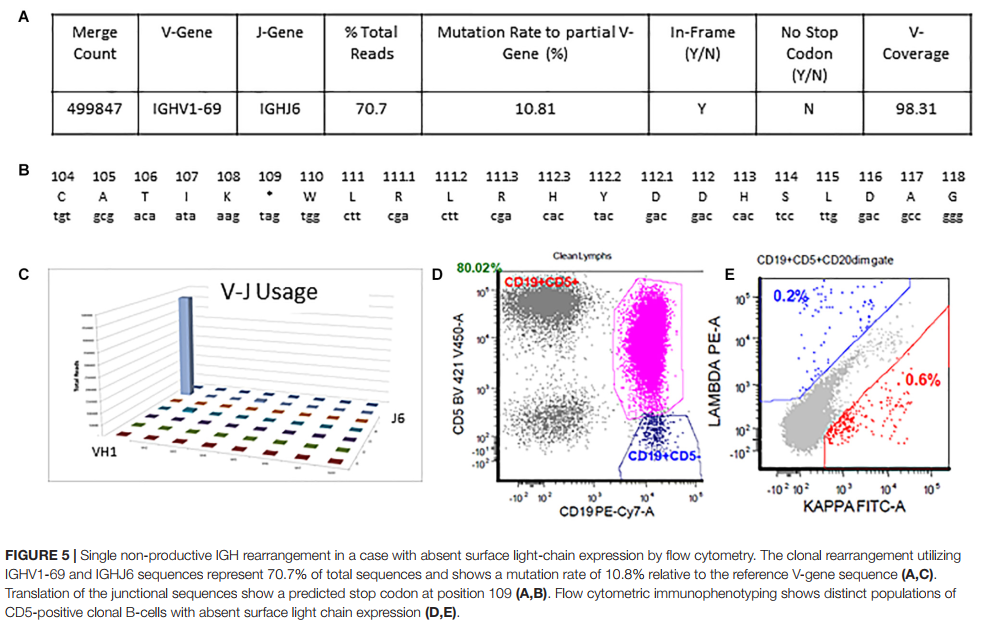
4.2 情况2:单个非生产性重排 Single Non-productive Rearrangement
情况2的详细输出可以在图5中看到。总读取计数为706770,而最小可报告读取计数为180,000(图5A,C)。 在这种情况下,存在一个克隆,该克隆具有非生产性重排,在核苷酸109位存在终止密码子。 该克隆占总读取的70.7%,使用V1-69和J6。 使用IMGT / HighV-QUEST和IgBLAST程序进行了手动交叉检查,以评估任何生产性重排,并确认了相同的发现(图5B)。 最终的解释是,由于克隆重排的非生产性状态,无法确定体细胞突变状态。 在流式细胞仪上,不存在支持IGHV重排的非生产性的表面轻链表达(图5D,E)。
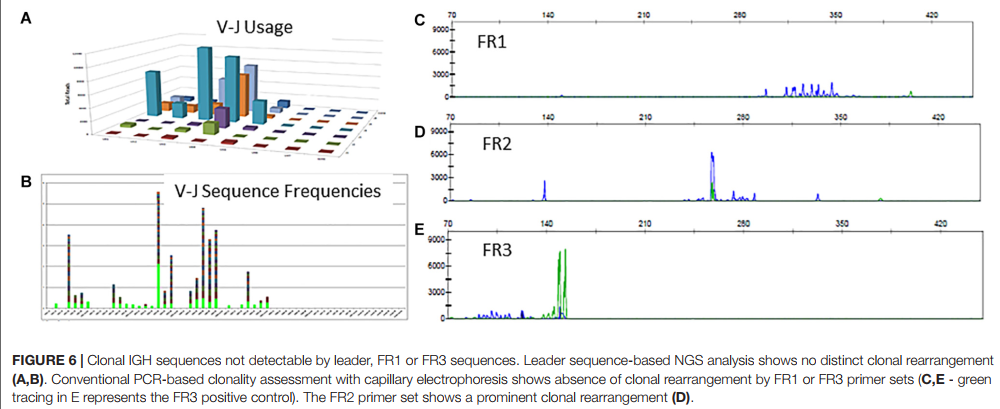
4.3 情况3:领导者或FR1引物无法检测到克隆重排 Clonal Rearrangement Not Detectable by Leader or FR1 Primers
在Lymphotrack数据输出的可视显示中,存在具有几个显性克隆的多克隆模式(图6A,B)。 IGH克隆性分析的分析表明,该模式仅使用FR2引物是克隆的(图6D),而使用FR1和FR3引物是多克隆的(图6C,E),这表明前导,FR1和FR3引物在这里失败,只有FR2引物 正在检测克隆重排。 因此,最终评估是由于前导引物的次佳扩增,无法确定体细胞突变状态。 当主要引物组未能检测到克隆重排时,此额外的分析和文档与ERIC建议使用独立引物组进行分析的建议一致。
五、未来发展方向
需要制定共识验证指南,实践标准和特定的质量保证措施,以支持临床实验室中越来越多地采用基于NGS的SHM测试。如Stamatopoulos B.等人所见,NGS分析可能会导致检测出更高比例的具有多个生产性IGHV亚克隆的患者。 (2017)。由于NGS的灵敏度较高,因此可以预期。而且,与Sanger测序相比,NGS具有更好的扩增和可靠测序DNA模板混合物的能力。然而,使用更敏感的NGS技术检测多个克隆和亚克隆挑战了淋巴瘤形成中的“单克隆性”传统概念,这意味着定义这些其他数据和发现的临床意义面临新挑战。希望这些新的数据解释方面可以在可测量的残留疾病检测,B细胞IG受体定型分析和了解CLL患者的疾病转化方面增加临床应用。
参考资料
- Front Cell Dev Biol. 2020; 8: 357.doi: 10.3389/fcell.2020.00357。 Evaluation of Somatic Hypermutation Status in Chronic Lymphocytic Leukemia (CLL) in the Era of Next Generation Sequencing 。 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7248390/
