【7.5】表位受体的结构复合物数据库( SCEptRe )
官网: http://tools.iedb.org/sceptre
准确的表位预测工具的开发对于促进疾病的诊断,治疗和疫苗开发非常重要。利用抗体和TCR序列信息来预测受体特异性表位的新方法的出现具有改变表位预测领域的潜力。这些新一代的表位预测方法的开发和验证,将受益于定期更新的高质量受体-抗原复合物数据集。
为了满足对高质量数据集的需求,以基准化这些新一代的受体特异性表位预测工具的性能,创建了一个名为 SCEptRe (Structural Complexes of Epitope-Receptor)的网络服务器。SCEptRe每周从免疫表位数据库中提取抗体-抗原,TCR-pMHC和MHC-配体的每周更新3D复合物,并根据抗原,受体和表位特征将它们聚类以生成基准数据集。SCEptRe还提供注释信息,例如受体上的CDR序列和VDJ基因。用户可以根据需要通过选择结构质量阈值和聚类参数(例如分辨率,R-free factor,抗原或表位序列同一性)来生成自定义数据集。
SCEptRe提供免疫受体-表位结构复合物的每周更新的,用户定制的全面基准数据集。这些数据集可用于将来开发和基准化受体特异性表位预测工具的性能。可从 http://tools.iedb.org/sceptre 免费访问SCEptRe。
一、背景
B和T细胞反应是适应性免疫的重要组成部分,可以为各种病原体提供长期保护。抗体和T细胞受体(TCR)分别由B细胞和T细胞表达,以识别不断变化的抗原集合。抗体和TCR识别抗原的特定区域(称为表位 epitope)及其结合位点(称为互补位 paratope)。对于许多医学,免疫和生物学应用,包括疾病控制,诊断和疫苗开发,表位的鉴定非常重要[1、2]。
表现最好的抗原序列和基于结构的B细胞表位预测工具,例如BepiPred,DiscoTope,ABCpred和CBtope [3,4,5,6],具有有限的预测能力[4]。这些B细胞表位预测工具可预测抗原上的表面斑块,该表面斑块可能是来自宿主的数十亿种抗体中的一种或多种抗体的靶标。鉴于这种庞大的抗体库,大多数抗原表面斑块可以成为宿主抗体的靶标,并且该特性是B细胞表位预测方法性能不佳的主要原因之一[7]。相反,T细胞表位的预测主要依赖于MHC结合预测[8,9,10]。 MHC结合是必要的,但不足以诱导免疫反应。需要适当的可以识别特定 peptide-MHC (pMHC) 复合物的T细胞克隆来诱导免疫反应。
免疫受体库测序(immune receptor repertoire )的最新进展[11]引起了人们对通过预定抗体和TCR识别表位的兴趣。因此,新一代的B和T细胞表位预测方法已经将重点从预测抗原中的一般表位转移到了预测特定受体的表位[12,13]。最近,几种抗体和TCR特异性表位的预测方法已经可用[7,13,14,15]。当前,训练和验证这些方法所需的数据很少,而且往往是重复的。受体特异性表位预测方法利用不同的聚类方法来消除其训练和测试数据集中的冗余,这使得难以可靠地比较和评估来自多种预测方法的结果。还需要评估这些方法在独立数据集上的性能。然而,由于用于训练和开发不同工具的方法和/或数据集尚不完全可用,因此对此类独立数据集的定义通常并非易事。
免疫抗原决定簇数据库(IEDB,Immune Epitope Database )是一个免费的公共资源,可捕获实验性免疫抗原决定簇和特定于抗原决定簇的受体数据,这些数据均由人工策划[16,17]。虽然存在包含抗体和TCR信息的其他3D结构数据库,[18,19,20],但IEDB将这些数据与所有其他种类的表位作图实验整合在一起,并包括针对每个表位识别的表位的标准化定义,包括对每个表位的手动质量检查每个数据元素,并允许用户批量下载数据库。截至2019年6月,IEDB拥有来自20,300多个手动策划参考中的585,000多个表位。 IEDB还为受体-抗原复合物的3D结构提供了计算的分子间接触和界面面积。受体-抗原复合物的这种原子级细节对于我们理解免疫受体的表位识别机制很重要。
为了满足对高质量数据集进行基准测试特定受体表位预测工具性能的需求,开发了一种称为 SCEptRe(表位受体的结构复合体)的网络服务器。基于抗原,表位和受体序列和/或结构特征,使用层次聚类来开发受体-表位复合物的全面非冗余数据集。
二、结果
2.1 IEDB中的受体-抗原复合物
IEDB用于提取表征免疫受体-抗原3D复合物的实验。截至2019年6月,在IEDB中总共提取了2510种Ab-Ag,296种TCR-pMHC和1107种MHC-配体复合物。这些复合物包括319种Ab-Ag,115种TCR-表位-MHC和147种MHC-配体与非肽类抗原的复合物,将在最后进行进一步讨论。对于肽表位,如以下各节所述,根据分辨率,抗原或线性表位序列长度以及CDR区中缺失残基的默认阈值,进一步过滤受体-抗原3D复合物。
2.2 Ab-Ag数据集
在所有2510种Ab-Ag复合物中
- 使用默认的3Å阈值对来自IEDB的总共2191种具有肽表位的Ab-Ag复合物进行过滤,以去除较差的分辨率结构。
- 还从数据集中删除了具有少于50个残基且抗体CDR区域中缺少残基的抗原的Ab-Ag复合物。
- 基于它们的抗原序列同一性,抗体CDR序列和表位3D构象相似性,将过滤后的772个Ab-Ag复合物中的聚类。使用70%序列同一性阈值将抗原序列独立聚类为338个不同的组。
- 根据所有可用抗体的CDR序列将其分为479个不同的组,并使用PocketMatch [21]基于3D构象相似性将其相应的表位进一步分为727个不同的抗体表位组(表1)。
2.3 TCR-pMHC数据集
从具有肽表位的IEDB的181个TCR-pMHC复合物的数据集中,使用3Å分辨率阈值去除了低分辨率复合物。还从数据集中滤出了核心表位少于8个残基的TCR-pMHC复合物和残基缺失的CDR。基于它们的核心-表位序列相似性,CDR和MHC G结构域序列,将过滤后的总共154个TCR-pMHC复合物进一步聚类为105个不同的簇,包括98个不同的TCR-表位基团(表1)。这些簇分别包括63、79和33个不同的T细胞核心表位,TCR和MHC分子组。
2.4 MHC-肽数据集
与TCR-pMHC复合物相似,没有考虑将MHC-肽复合物分离度大于3Å,并且残基核心表位小于8个。在960种MHC肽复合物中,过滤后剩余的861种MHC肽复合物根据其核心肽序列相似性和MHC G结构域序列进一步聚类为439个不同的簇(表1)。这些簇分别包括342和133个不同组的核心肽和MHC分子。
2.5 非肽表位或配体数据集 Non-peptidic epitope or ligand datasets
基于抗体CDR区域中的分辨率和缺失残基,总共过滤了具有非肽表位的319种Ab-Ag复合物。 根据其CDR序列,将281种Ab-Ag复合物的其余抗体序列分为154个不同的抗体组。 类似地,所有基于非肽配体的115种TCR-pMHC复合物均基于3Å分辨率阈值进行了过滤,并且在CDR区域中没有缺失的残基。 将其余的85个TCR-pMHC复合物聚类为28个TCR和7个MHC不同的组。 基于其对126种复合物的分离度,共过滤了147种具有非肽类配体的MHC-配体复合物。 根据其MHC序列将这126个MHC-配体复合物分为18个不同的组。
2.5 网络服务器
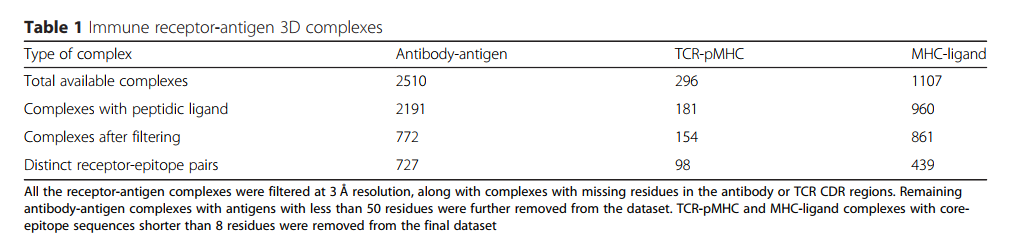
开发了一个名为SCEptRe(表位-受体的结构复合体)的网络服务器,并在IEDB分析资源中提供[22],以提供每周更新的受体-抗原3D复合体(图1)。可以在 http://tools.iedb.org/sceptre 中访问SCEptRe。所有结构质量参数,例如分辨率,用于过滤受体-抗原复合物的CDR中缺失的残基,都可以通过SCEptRe获得。此外,基于R自由因子的过滤已添加到Web服务器。还提供了抗原和表位/配体过滤和聚类参数。此外,还向用户提供了MHC特征参数,即TCR-pMHC和MHC-配体3D复合物的来源生物和MHC类。还选择了带有肽或非肽表位/配体的受体-抗原复合物的选项。 “方法”部分中讨论的所有默认过滤和聚类参数均作为在线Web服务器中的建议值提供,用户可以在其中根据需要对其进行修改。
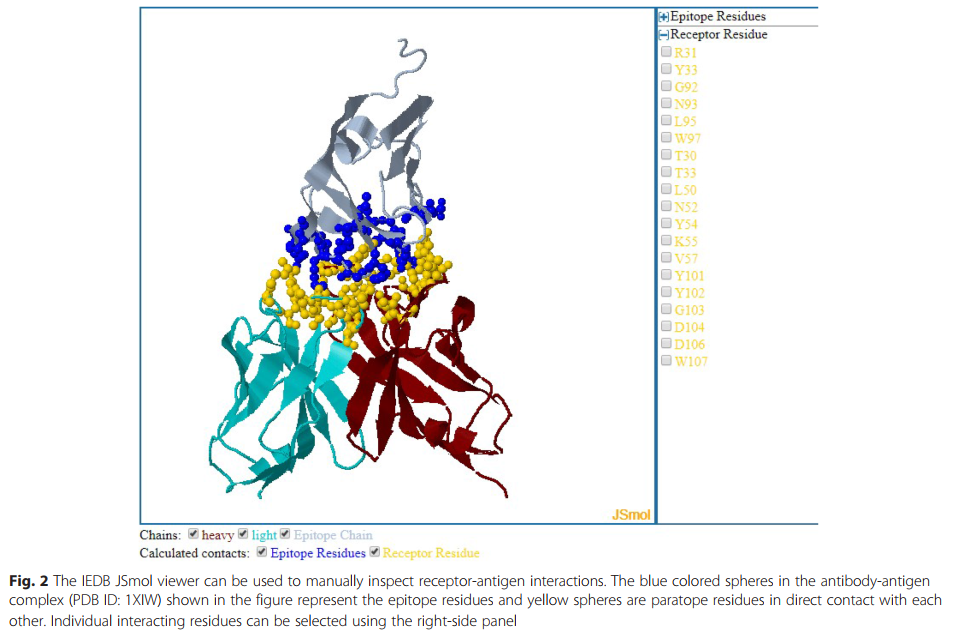
结果显示为表格,其中包含指向IEDB分析,表位和受体详细信息页面的链接,以及指向PDB网站的链接。 受体-抗原复合物的3D结构可以使用IEDB JSmol查看器(JSmol:用于3D化学结构的开放源代码HTML5查看器)进行可视化。 http://wiki.jmol.org/index.php/JSmol )(图2)提供检查受体-抗原复合物中一个或所有相互作用残基的选择。
三、讨论区
测序个人B细胞和T细胞受体库的技术的进步,导致了受体特异性B细胞和T细胞表位预测方法的发展。 这些新的表位预测方法将从定期更新的高质量受体-抗原数据集中受益。 IEDB之类的资源提供了有关抗原受体复合物实验信息的人工整理数据。 SCEptRE提供了几个下游处理步骤,以过滤来自IEDB的数据,以保留高质量的受体-抗原复合物。 SCEptRE还提供了将受体-抗原3D复合物聚类的方法,因此这些数据集可直接用于训练和验证受体特异性表位预测方法。
当前可用的3D受体-抗原复合物数据集基于抗体和TCR的全长序列对它们进行聚类[7、13、23、24]。 具有相同CDR的抗体和TCR序列可以通过这种方法分为不同的簇。 CDR与抗原直接相互作用,并与B细胞和T细胞受体的特异性有关[25,26]。 还已知抗体重链和TCR β链与抗原接触最广泛。 因此,SCEptRe会根据其CDR序列对受体进行分类。 受体簇标识符包括有关单个重链,轻链,α链或β链簇的信息,以便用户可以根据他们的需求(例如, 仅重链或仅β链基团。
SCEptRe还根据目前可用的抗体-抗原复合物数据集中的3D构象相似性,解决了B细胞表位缺乏分组的问题。 与同源抗原的构象相似的表位结合的交叉反应抗体可以提供相似的Ab-Ag接触残基对。 这样的Ab-Ag复合物可以通过它们的表位之间的构象相似性分组在一起。
一年来,科学界已使用不同的MHC等位基因命名法来描述MHC分子。 SCEptRe引入了基于序列的方法,对TCR-pMHC和MHC-配体复合物应用MHC分子的一致分类,而不是依靠报道的MHC分子鉴定提供一致的MHC分子簇。
四、结论
SCEptRe提供用户定制的每周更新的受体-抗原3D复合物非冗余数据集以及带注释的受体信息。 SCEptRe提供的数据集可用于将来开发和基准化受体特异性表位预测工具的性能。 受体-抗原复合物的聚类可用于从表位预测算法的训练和验证数据集中删除冗余。
五、方法
5.1 数据集
设置了一个自动查询,该查询从IEDB SQL数据库中提取了抗体抗原,TCR-pMHC和MHC-配体分析,该数据库具有3D结构。检索与宿主生物,源抗原,表位,受体的CDR序列和VDJ基因有关的信息,以及PDB ID,分辨率和计算出的受体-抗原接触(使用4Å距离)。在IEDB策划过程中手动检查计算出的受体-抗原接触,以检查和去除不相关的接触,这些接触不是抗原-受体相互作用的一部分,尤其是在工程抗体,TCR或MHC构建体的情况下。从其 PDBx / mmCIF文件中提取复合物的无R因子,并将其添加到SQL查询输出文件中。此外,将所有受体-抗原3D复合物过滤以去除不良的结构。具有肽抗原的受体-抗原复合物(包括全长蛋白,但不包含非肽小分子)根据其受体和抗原特征进一步聚类,以识别不同的受体-表位对。相反,具有非肽表位的受体-抗原复合物根据它们的受体特征分别聚类。
5.2 抗体-抗原(Ab-Ag)复合物
抗体链序列使用ANARCI进行编号[27],CDR使用IMGT编号进行标识[28]。将抗体CDR序列映射到其3D结构。筛选以排除包括的低质量结构,分辨率(默认值:3Å)和可选的CDR区中缺少骨架原子的复合物的去除(默认值:remove)。具有较短抗原蛋白序列的Ab-Ag复合物可以进一步过滤(默认值:排除具有<50个氨基酸残基的抗原)。
使用具有序列同一性阈值(默认值:70%)的聚类工具[29],根据其抗原序列将其余的Ab-Ag复合物聚类。此外,基于抗体CDR序列将所有Ab-Ag复合物聚类。具有相同CDR序列的抗体重链和轻链被独立分组,它们的簇分别用“ H”和“ L”前缀表示。例如,抗体簇H1_L1,其中H1是第一个重链簇,L1是第一个轻链簇。
PocketMatch(版本2)用于鉴定构象相似的B细胞表位[21]。 PocketMatch计算2个结合位点中所有原子距离对的所有值,然后比较距离的排序列表以识别构象相似的结合位点。 PocketMatch提供两种类型的分数,即PMax和PMin。 PMax分数是两个结合位点中匹配的距离对与更长结合位点中距离对总数的比值。 相反,PMin得分是2个结合位点中匹配的距离对与较短结合位点中的距离对总数之比。 PMax和PMin得分均用于将B细胞表位聚类在每个抗体簇内,以产生独特的抗体-表位对。 如果它们的PMax或PMin评分为1.0,或者PMin评分至少为0.9,PMax评分至少为0.6,则来自每个抗体簇的成对B细胞表位会聚在一起。
5.3 TCR-pMHC复合物
像Ab-Ag复合物一样,CDR序列是通过使用IMGT编号的ANARCI对TCR链序列编号来识别的。根据分辨率(默认值:3Å)和CDR区域中缺少骨架原子的情况(默认值:remove)对TCR-pMHC复合物进行过滤,以排除低质量的结构。另外,定义核心表位以忽略不直接与MHC或TCR分子相互作用的抗原肽的突出端。核心表位定义包括在MHC或TCR分子中任何原子4Å距离之内的抗原链上第一个和最后一个残基之间的所有残基。从数据集中删除了核心表位序列短于定义的残基数(默认值:8个残基)的TCR-pMHC复合物。
使用簇工具基于序列表位阈值为(默认值:85%)的核心表位序列对剩余的TCR-pMHC进行聚类。将具有相同序列的TCRα和β链聚类。像抗体聚类一样,TCR分别基于其α和β链CDR进行聚类,并分别以“ A”和“ B”前缀表示。
MHC I类和II类分子中的α1-α2和α1-β1结构域分别称为凹槽或G结构域[30]。 MHC分子的配体结合位点是G结构域的一部分,因此负责其特异性。从IMGT [31]下载了所有可用的G结构域序列。使用BLAST针对这些G结构域数据集搜索了来自TCR-pMHC复合体的MHC链序列[32]。 BLAST参数(例如e值为1.00E-10,查询覆盖率阈值为90%和主题覆盖率阈值为85%)用于将输入的MHC序列与已知的G域进行比对。提取映射到第一个G结构域命中的MHC序列区域,并用于聚类TCR-pMHC复合物。具有相同α1-α2结构域的MHC I类分子聚在一起,并用前缀“ a”表示。同样,具有相同α1和β1结构域的MHC II类分子也聚集在一起,并分别以“ a”和“ b”前缀表示。对于非经典MHC分子(例如人CD1a),将其完整蛋白质序列用于聚类,并使用“ n”作为前缀表示基团。
5.4 MHC-肽复合物
MHC-肽复合物的核心肽定义为在MHC分子中任何原子的4Å距离之内,抗原链上第一个和最后一个残基之间的残基范围。 从数据集中删除了核心肽短于8个残基的MHC肽复合物。 MHC-肽复合物使用序列同一性阈值(默认值:85%)基于核心肽聚类,就像TCR-pMHC复合物中的核心表位一样。 此外,这些复合物基于MHC G结构域进行聚类,就像TCR-pMHC复合物中的MHC G结构域序列聚类一样。
5.5 与非肽抗原的复合物
根据分辨率(默认值:3Å)和CDR区中缺失残基的存在(默认值:remove)过滤所有具有非肽表位的复合物。 如适用,如前面所述,其余的复合物根据其受体CDR序列和MHC分子进行聚类。
参考资料
- Mahajan, S., Yan, Z., Jespersen, M. C., Jensen, K. K., Marcatili, P., Nielsen, M., Sette, A., Peters, B. 2019. Benchmark datasets of immune receptor-epitope structural complexes. BMC Bioinformatics , 20(1), 490. 网址: https://doi.org/10.1186/ https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3109-6
