【4.3.1.3】T细胞和B细胞表位预测的基本原理和方法
摘要
适应性免疫(Adaptive immunity)由T细胞和B细胞介导,T细胞和B细胞是能够产生赋予免疫保护的病原体特异性记忆的免疫细胞。 B细胞和T细胞的记忆和效应功能基于通过病原体(pathogens)中特定靶标(抗原)的特异性受体的识别来预测。更具体地,B细胞和T细胞识别其同源抗原中称为表位(epitopes)的部分。
出于许多实际原因,人们对鉴定抗原中的表位非常感兴趣,包括了解疾病病因学,免疫监测,开发诊断测定和设计基于表位的疫苗。表位鉴定是昂贵且耗时的,因为它需要对大量潜在表位候选物进行实验筛选。幸运的是,研究人员已经开发出计算机预测方法,通过减少用于实验测试的潜在表位候选者列表,显着减少与表位作图相关的负担。
- 我们分析与表位预测相关的T细胞和B细胞的抗原识别方面。
- 我们对最相关的B细胞和T细胞表位预测方法和工具进行系统和包容性的综述。
一、介绍
免疫系统通常分为两类: 先天性 ( innate )和自适应性 ( adaptive )。 先天免疫涉及非特异性防御机制,其在体内微生物出现后立即或数小时内起作用。 所有多细胞生物都表现出某种先天免疫力。 相反,适应性免疫仅存在于脊椎动物中,并且具有高度特异性。 事实上,适应性免疫系统能够单独识别和摧毁入侵的病原体。 此外,适应性免疫系统记住了战斗的病原体,获得了病原体特异性的持久保护记忆,每次病原体再次发生时都会产生更强的攻击[1]。 尽管如此,先天性和适应性免疫机制共同作用,适应性免疫诱导依赖于先天免疫应答的先前激活
适应性免疫由淋巴细胞表达,更具体地由B细胞和T细胞表达,其负责体液和细胞介导的免疫。 B细胞和T细胞不能识别病原体(pathogens)整体,而是称为抗原(antigens)的分子成分。 这些抗原被B细胞和T细胞的细胞表面中存在的特异性受体识别。 这些受体的抗原识别是激活B细胞和T细胞所必需的,但还不够,因为还需要源自先天免疫系统激活的第二激活信号。 识别的特异性由淋巴细胞发育过程中发生的遗传重组事件(genetic recombination events)决定,这导致在抗原识别受体方面产生数百万种不同的淋巴细胞变体[1]。 B细胞和T细胞的抗原识别差别很大
B细胞通过抗原受体识别溶剂暴露的抗原,称为B细胞受体(B-cell receptors,BCR),由膜结合的免疫球蛋白组成,如图1所示。激活后,B细胞分化并分泌可溶形式的免疫球蛋白, 也称为抗体,其介导体液适应性免疫。 由B细胞释放的抗体可以具有在结合其同源抗原时触发的不同功能。 这些功能包括中和毒素( neutralizing toxins)和病原体并将其标记为破坏[1]。
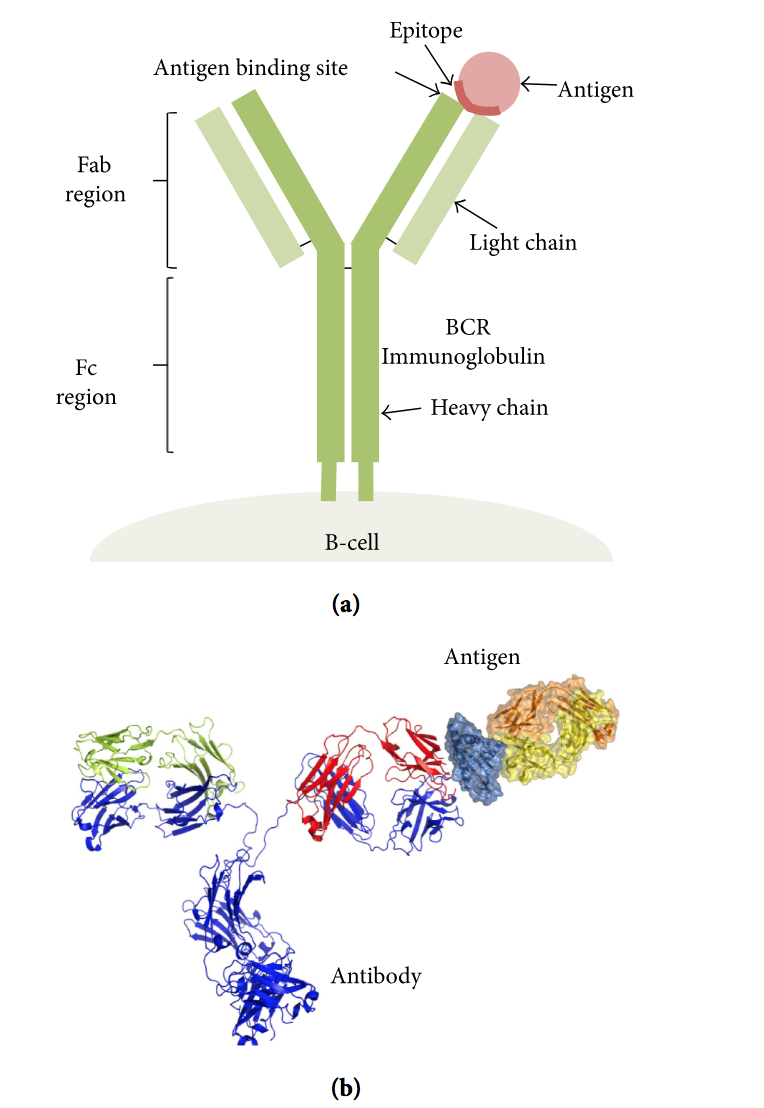
图1:B细胞表位识别。 B细胞表位是抗原的溶剂暴露部分,其与分泌的和细胞结合的免疫球蛋白结合。 (a)B细胞受体(B-cell receptors )包括细胞结合的免疫球蛋白,由两条重链和两条轻链组成。 不同的链和区域都有注释。 (b)抗体与抗原之间相互作用的分子代表。 抗体是已知特异性的分泌型免疫球蛋白。
B细胞表位(B-cell epitope)是结合免疫球蛋白或抗体的抗原部分。 B细胞识别的这些表位可以构成抗原中任何暴露的溶剂区域,并且可以具有不同的化学性质。 然而,大多数抗原是蛋白质,并且那些是表位预测方法的主题。
另一方面,T细胞在其表面存在一种称为T细胞受体(T-cell receptor,TCR)的特异性受体,当它们显示在与主要组织相容性复合物(MHC)分子结合的抗原呈递细胞(APC)表面上时能够识别抗原。 。 T细胞表位由I类(MHC I)和II(MHC II)MHC分子呈现,其分别被两个不同的T细胞亚群,CD8和CD4 T细胞识别(图2)。 随后,存在CD8和CD4 T细胞表位。 在T CD8表位识别后,CD8 T细胞变成细胞毒性T淋巴细胞(CTL)。 同时,引发的CD4 T细胞成为辅助(Th)或调节(Treg)T细胞[1]。 Th细胞扩增免疫反应,有三个主要的亚类:Th1(细胞介导的针对细胞内病原体的免疫),Th2(抗体介导的免疫)和Th17(炎症反应和对细胞外细菌的防御)[
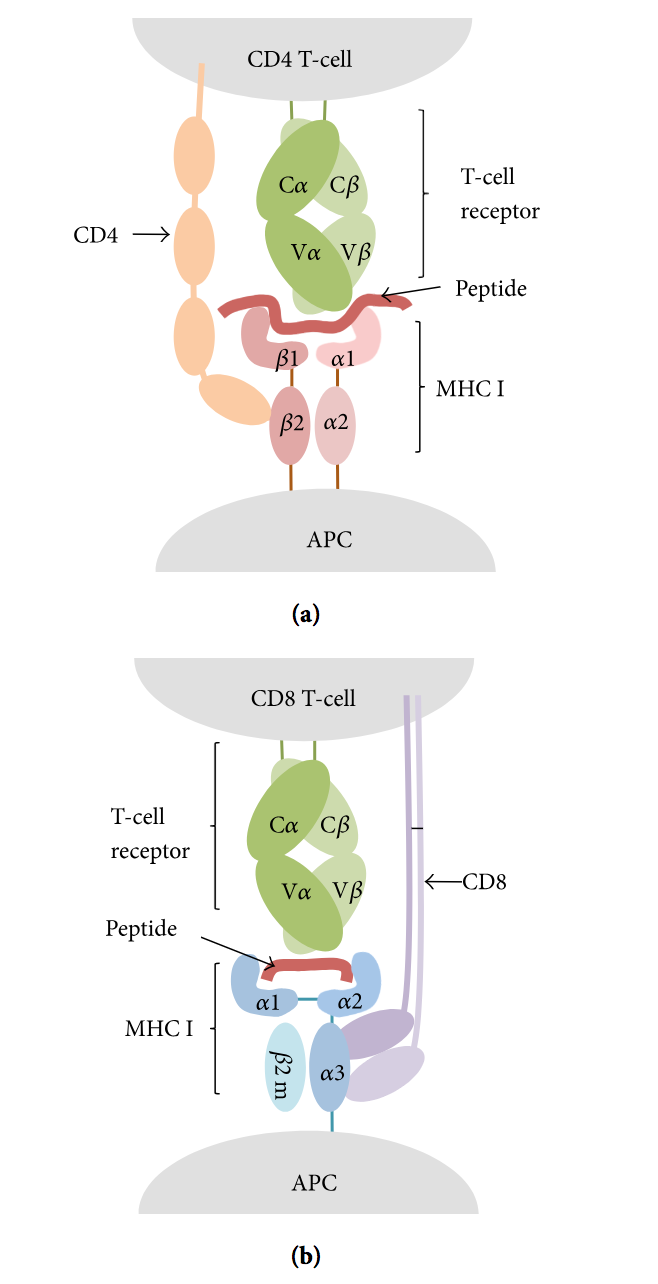
图2:T细胞表位识别。 T细胞表位(T-cell epitopes )是衍生自抗原的肽,并且当与APC的细胞表面上展示的MHC分子结合时被T细胞受体(T-cell receptor,TCR)识别。 (a)CD4 T细胞表达CD4辅助受体,其结合MHC II,并识别由MHC II分子呈递的肽。 (b)CD8 T细胞表达CD8辅助受体,其结合MHC I,并识别由MHC I分子呈递的肽。 (这个peptiede来自抗原哦)
由于许多实际原因,包括了解疾病病因学,免疫监测,开发诊断测定和设计基于表位的疫苗,鉴定抗原中的表位是非常令人感兴趣的。 B细胞表位可以通过不同的方法鉴定,包括解决抗原 - 抗体复合物的3D结构,抗体结合的肽文库筛选或进行抗原突变的功能测定,并评估相互作用抗体 - 抗原[3,4] 。另一方面,T细胞表位的实验测定使用MHC多聚体和淋巴细胞增殖或ELISPOT测定等[5,6]进行。传统的表位鉴定完全取决于实验技术,昂贵且耗时。因此,科学家已经开发并实施了表位预测方法,其促进表位鉴定并减少与其相关的实验负荷。在这里,我们将首先分析T细胞和B细胞的抗原识别方面,这些方面与更好地理解表位预测的主题相关。随后,我们将对最重要的预测方法和工具进行系统和包容性的审查,特别注意其基础和潜力。我们还将讨论表位预测的局限性和克服它们的方法。我们将从T细胞表位开始。
二、T-Cell Epitope Prediction
T细胞表位预测旨在鉴定抗原内能够刺激CD4或CD8 T细胞的最短肽[7]。 这种刺激T细胞的能力被称为免疫原性(immunogenicity),并且在需要衍生自抗原的合成肽的试验中证实[5,6]。 抗原内有许多不同的肽,T细胞预测方法旨在鉴定那些具有免疫原性的肽。 T细胞表位免疫原性(T-cell epitope immunogenicity)取决于三个基本步骤:
- 抗原加工 antigen processing
- 肽与MHC分子的结合
- 同源TCR识别 (recognition by a cognate TCR)
在这三个事件中,MHC-肽结合是决定T细胞表位的最具选择性的[8,9]。 因此,预测肽-MHC结合是预测T细胞表位的主要基础,我们接下来将对其进行回顾。
2.1 肽-MHC结合的预测 Prediction of Peptide-MHC Binding
MHC I和MHC II分子具有相似的3D结构,其中结合的肽位于由两个α-螺旋描绘的凹槽中,所述两个α-螺旋覆盖由八个反平行β-链组成的底板。然而,MHC I和II结合沟之间也存在关键差异,我们必须强调它们是条件肽结合预测(图3)。 MHC I分子的肽结合裂缝是封闭的,因为它是由单个α链构成的。因此,MHC I分子只能结合9到11个氨基酸的短肽,其N-末端和C-末端仍然通过氢键网络固定在MHC I分子的保守残基上[10,11]。 MHC I肽结合沟还含有深度结合口袋,具有紧密的物理化学偏好,有助于结合预测。然而,有一个复杂的问题。具有不同大小并与相同MHC I分子结合的肽通常使用替代的结合口袋[12]。因此,预测肽-MHC I结合的方法需要固定的肽长度。然而,由于大多数MHC I肽配体具有9个残基,因此通常优选预测具有该大小的肽。相反,MHC II分子的肽结合沟是开放的,允许肽的N-和C-末端延伸超过结合沟[10,11]。结果,MHC II结合的肽的长度变化很大(9-22个残基),尽管只有9个残基的核心(肽结合核心)位于MHC II结合沟中。因此,肽-MHC II结合预测方法通常靶向鉴定这些肽结合核心。 MHC II分子结合口袋也比MHC I分子更浅且要求更低。因此,对MHC II分子的肽结合预测不如MHC I分子准确。
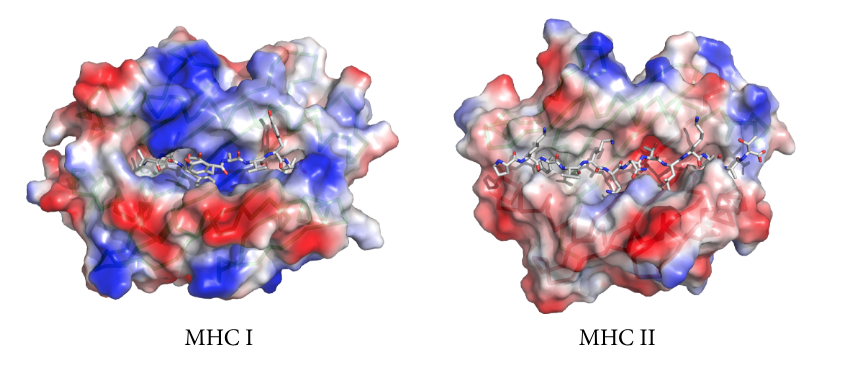
图3:MHC分子结合沟(molecule binding groove)。 该图描绘了代表性MHC I和II分子的TCR所见的分子表面。 注意MHC I分子的结合沟是如何闭合的,而MHC II的结合沟是如何打开的。 结果,MHC I分子结合短肽(8-11个氨基酸),而MHC II分子结合更长的肽(9-22个氨基酸)。 该图是使用PyMol从PDB文件1QRN(MHC I)和1FYT(MHC II)制备的。
鉴于该问题的相关性,有许多方法来预测肽-MHC结合。 表1收集了与免费在线使用最相关的内容。它们可分为两大类:数据驱动(data-driven )和基于结构(structure-based)的方法。 基于结构的方法通常依赖于对肽-MHC结构的建模,然后通过诸如分子动力学模拟的方法评估相互作用[8,13]。 基于结构的方法具有不需要实验数据的巨大优势。 然而,它们很少被使用,因为它们是计算密集型的并且表现出比数据驱动方法更低的预测性能[14]。
用于肽-MHC结合预测的数据驱动方法是基于已知与MHC分子结合的肽序列。这些肽序列通常可在专门的表位数据库中获得,例如IEDB [15],EPIMHC [16],Antijen [17,18]。 MHC I和II结合肽都在特定肽位置含有频繁出现的氨基酸,称为锚定残基(anchor residues)。因此,首先使用反映锚定位置MHC分子的氨基酸偏好的序列基序(sequence motif ,SM)来预测肽-MHC结合[19]。然而,很快就发现非锚定残基也有助于肽与给定MHC分子结合的能力[20,21]。随后,研究人员开发了基序矩阵(motif matrices,MM),可以评估每个和所有肽位置对MHC分子结合的贡献[22-25]。最复杂的基序矩阵形式由[24-26]组成,与检测序列同源性相似[27]。我们想要注意的是,基序矩阵经常与定量亲和基质(quantitative affinity matrices,QAM)混淆,因为它们都产生肽分数。然而,在不考虑结合亲和力的值的情况下得到MM,因此,得到的肽得分不适于解决结合亲和力。相反,QAM在肽和相应的结合亲和力上进行训练,并且旨在预测结合亲和力。基于QAM的第一种方法是由Parker等人开发的。 [28](表1)。随后,开发了各种方法以从肽亲和力数据获得QAM并预测肽与MHC I和II分子的结合
QAM和基序矩阵可能对肽侧链对结合具有独立贡献。 实验数据很好地支持了这一假设,但也有证据表明邻近的肽残基会干扰其他肽[33]。 为了解释这些干扰,研究人员引入了定量构效关系(quantitative structure-activity relationship ,QSAR)加性模型,其中肽与MHC的结合亲和力计算为每个位置的氨基酸贡献总和加上相邻侧链相互作用的贡献[34]。 然而,机器学习(ML)是最常用和最强大的方法,用于处理肽-MHC结合数据的非线性[8]。 研究人员使用ML来解决两个不同的问题:MHC结合剂与非结合的区分以及肽对MHC分子的结合亲和力的预测。
为了开发鉴别模型,ML算法在由肽结合的数据集上训练,包含肽结合或不结合MHC分子。基于ML的辨别模型的相关示例是基于人工神经网络(ANN)[35,36],支持向量机(SVM)[37-39],决策树(DT)[40,41]和隐马尔可夫的模型(HMM),他们可以处理非线性数据,并已用于区分与MHC分子结合的肽。但是,与其他ML算法不同,它们必须仅针对正数据进行训练。已经使用三种类型的HMM来预测MHC-肽结合:完全连接的HMM(fully connected HMMs) [42],结构优化的HMM(structure-optimized HMMs) [43]和分布HMM(profile HMMs) [,43,44]。其中,只有完全连接的HMM(fcHMM)和结构优化的HMM(soHMM)才能识别肽结合物中的不同模式。实际上,衍生自无空位比对组的profile HMMs(结合MHC的肽的情况)几乎与分布矩阵相同[45]
关于预测结合亲和力,ML算法在由对MHC分子具有已知亲和力的肽组成的数据集上训练。 SVM和ANN都用于此目的。 首先应用SVM预测与MHC I分子的肽结合亲和力[46],然后预测MHC II分子[47](表1)。 同样,人工神经网络也首先应用于预测肽与MHC I的结合[48,49],然后应用于MHC II分子[50](表1)。 肽-MHC结合预测方法的基准似乎表明基于ANN的那些优于基于QAM和MM的那些。 然而,不同方法之间的差异是微不足道的,并且因不同的MHC分子而异[51]。 此外,已经表明通过组合几种方法并提供共识预测来改善肽-MHC预测的性能[52]。
通过肽-MHC结合模型预测T细胞表位的主要难度是MHC多态性。 在人类中,MHC分子被称为人白细胞抗原(HLA),并且存在数百种I类(HLA I)和II类(HLA II)分子的等位基因变体。 这些HLA等位基因变体结合不同的肽组[53],需要特定的模型来预测肽-MHC的结合。 然而,肽结合数据仅适用于少数HLA分子。 为了克服这一局限性,一些研究人员通过对输入数据进行人工神经网络训练,结合MHC残基与肽结合亲和力,能够预测与未鉴定的HLA等位基因的肽结合亲和力,开发了泛MHC特异性方法[54,55]。
HLA多态性也阻碍了全世界覆盖基于T细胞表位的疫苗的发展,因为HLA变体在不同种族群体中以可变很大的频率[56]。有趣的是,不同的HLA分子也可以结合相似的肽组[57,58],研究人员已经设计出将它们分组的方法,称为HLA超型(supertypes),由具有相似肽结合特异性的HLA等位基因组成[59-61]。 HLA-A2,HLA-A3和HLA-B7是超型的相关实例; 88%的样本至少表达了这些超类型中包含的等位基因[25,57,58]。鉴定混杂的肽与HLA超型的结合,使得能够使用有限数量的肽开发具有高群体覆盖的T细胞表位疫苗。目前,一些基于网络的方法允许预测混杂的肽结合HLA超型用于表位疫苗设计,包括MULTIPRED [62]和PEPVAC [63](表1)。 Molero-Abraham等人开发并实施了鉴定超出HLA超型的混杂肽结合的方法 [64]名称为EPISOPT。 EPISOPT预测单个肽的HLA I呈递谱,无论超类型如何,并鉴定表位组合,提供更广泛的群体保护覆盖。
肽与MHC II分子结合的预测容易区分CD4 T细胞表位,但不能说明它们激活特定CD4 T细胞亚群(例如Th1,Th2和Treg)的反应的能力。 然而,有证据表明某些CD4 T细胞表位似乎刺激Th细胞的特定亚群[65,66]。 区分MHC II限制性表位引发不同反应的能力显然与表位疫苗的发展相关,并引起了研究人员的注意。 一个相关的例子是Dhanda等人的工作[67],通过在实验验证的IL4诱导和非诱导MHC II类结合物上训练SVM模型,产生能够预测Th2细胞典型的白细胞介素4(IL-4)分泌的潜在肽诱导物的分类器(表1)。
2.2 通过肽-MHC结合预测抗原加工及整合(Antigen Processing and Integration)
抗原加工塑造了可用于MHC结合的肽库,并且是决定T细胞表位免疫原性的限制步骤[68]。 随后,抗原加工途径的计算建模提供了增强T细胞表位预测的手段。 MHC I和II分子的抗原呈递通过两种不同的途径进行。 MHC II分子呈递源自内吞抗原(endocyted antigens)的肽抗原,其被降解并加载到内体区室(endosomal compartments)中的MHC II分子上[69]。 II类抗原降解知之甚少,尚缺乏良好的预测算法[70]。 相反,MHC I分子呈现主要衍生自细胞溶质中降解的抗原的肽。 然后通过TAP将得到的肽抗原转运到内质网,在那里它们被加载到新生的MHC I分子上[69](图4)。 在上样之前,肽经常被ERAAP N-末端氨基肽酶修剪[71]

图4:I类抗原加工。 该图描绘了MHC I分子的抗原呈递中涉及的主要步骤。 蛋白酶(proteasome)体降解蛋白质,肽片段通过TAP转运至内质网(endoplasmic reticulum,ER),在那里它们被加载到新生的MHC I分子上。 TAP转运8至16个氨基酸的肽。 长肽不能结合MHC I分子,但经常变得适合于通过ERAAP进行N末端修剪后的结合。
已经详细研究了蛋白酶体切割和肽与TAP的结合,并且存在预测两种过程的计算方法。 蛋白酶体切割预测模型来源于人体组成型蛋白酶体体外产生的肽片段[72,73]和映射到其源蛋白上的MHC I限制性配体组[74-76]。 另一方面,TAP结合预测方法已经通过在已知亲和力的肽上训练不同的算法来开发[77-80]。 与仅与肽结合MHC I相比,蛋白酶体切割和肽与TAP的结合与肽-MHC结合预测相结合,增加了T细胞表位预测率[37,77,81-83]。 随后,研究人员开发了通过多步骤方法预测CD8 T细胞表位的资源,这些方法整合了蛋白酶体切割,TAP转运和肽与MHC分子的结合[26,37,82-85](表1)。
三、B细胞表位的预测
B细胞表位预测旨在促进B细胞表位鉴定,其实际目的是替换抗原用于抗体产生或用于进行结构功能研究。抗原中任何溶剂暴露的区域都可以被抗体识别。尽管如此,B细胞表位可分为两大类:线性和构象(linear and conformational )(图5)。线性B细胞表位由连续的残基,肽组成,而构象B细胞表位由来自残基的溶剂暴露的原子的片组成,其不一定是连续的(图5)。因此,线性和构象B细胞表位也分别称为连续和不连续的B细胞表位。识别线性B细胞表位的抗体可以识别变性抗原,而变性抗原导致对构象B细胞表位的识别丧失。大多数B细胞表位(约90%)是构象的,事实上,只有少数天然抗原含有线性B细胞表位[3]。我们将回顾线性和构象B细胞表位的预测。

图5:线性和构象B细胞表位。 线性B细胞表位(a)由连续/连续残基组成,而构象B细胞表位(b)沿序列包含分散/不连续残基。
3.1 线性B细胞表位的预测
线性B细胞表位由肽组成,这些肽可以用于替代引起免疫反映和抗体产生的抗原。因此,尽管是少数,但线性B细胞表位的预测已受到重视。使用基于序列的方法从抗原的一级序列预测线性B细胞表位。用于预测B细胞表位的早期计算方法基于描绘B细胞表位的物理化学特征的简单氨基酸倾向量表。例如,Hopp和Wood应用残基亲水性计算B细胞表位预测[96,97],假设亲水区主要位于蛋白质表面并具有潜在的抗原性。然而,我们现在知道蛋白质表面含有大致相同数量的亲水和疏水残基[98]。引入B细胞表位预测的其他氨基酸倾向量表(amino acid propensity scales),比如,灵活性[99],表面可及性[100]和β-转角倾向[101]。目前可用的生物信息学工具使用倾向量表来预测线性B细胞表位,包括PREDITOP [102]和PEOPLE [103](表2)。 PREDITOP [102]使用基于氨基酸的亲水性,可接近性,灵活性和二级结构特性的多参数算法。 PEOPLE [103]使用相同的参数,另外还包括β转弯的评估。 Kolaskar和Tongaonkar [104]介绍了一种预测B细胞表位的相关方法,该方法由一个简单的抗原性量表(antigenicity scale)组成,该抗原性量表来源于实验测定的B细胞表位中的物理化学性质和氨基酸频率。该指数可能是B细胞表位预测最常用的抗原量表,它实际上是由GCG [105]和EMBOSS [106]包实现的。在85个线性B细胞表位的数据集中进行的倾向量表的比较评估显示,大多数倾向量表预测50-70%的B细胞表位,β-转角量表达到最佳值[101,107]。已经表明,组合不同尺度似乎不会改善预测[102,108]。此外,Blythe和Flower [109]证明单尺度氨基酸倾向量表(single-scale amino acid propensity scales)对于预测表位位置是不可靠的。

表2:可用于免费在线使用的B细胞表位预测方法。
用于预测线性B细胞表位的氨基酸量表(amino acid scales)表现很差,促使引入基于机器学习(ML)的方法(表2)。这些方法是通过训练ML算法来开发的,以区分实验性B细胞表位和非B细胞表位。在训练之前,B细胞表位被翻译成捕获所选特性的特征向量,例如由不同倾向量表给出的特性。基于ML的B细胞表位预测方法的相关方法包括:BepiPred [110],ABCpred [111],LBtope [112],BCPREDS [113]和SVMtrip [114]。用于开发这些方法的数据集,训练特征和算法不同。 BepiPred基于从抗原 - 抗体复合物的3D结构获得的B细胞表位复合物,随机森林训练方法[110]。 BCPREDS [113]和SVMtrip [114]都基于支持向量机(SVM),但BCPREDS使用各种字符串内核进行训练,无需将序列表示为长度固定的特征向量,SMVtrip在长度固定的基础上进行了训练三肽(tripeptide)组成向量。 ABCpred和LBtope方法由人工神经网络(ANNs)组成,这些人工神经网络在相似的阳性数据,B细胞表位上进行训练,但在负数据,非B细胞表位上有所不同。用于训练ABCpred的负数据包括随机肽,而用于LBtope的负数据基于实验验证的非B细胞表位形成IEDB [15]。通常,据报道采用ML算法的B细胞表位预测方法优于基于氨基酸倾向量表的那些。尽管如此,一些作者报道ML算法与基于单尺度的方法相比几乎没有改进[115]。
在免疫应答过程中引发的抗体,通常具有决定其生物学功能的相同同种型(isotype)。 B细胞表位预测的最新进展是Gupta等人开发的方法 [116],这允许鉴定能够诱导特定类别抗体的B细胞表位。 该方法基于在数据集上训练的SMV,该数据集包括已知诱导IgG,IgE和IgA抗体的线性B细胞表位。
3.2 构象B细胞表位的预测
大多数B细胞表位是构象的(conformational),但是构象B细胞表位的预测已经落后于线性B细胞表位的预测。 这有两个主要的实际原因:
- 首先,构象B细胞表位的预测通常需要蛋白质三维(3D)结构的知识,而这些信息仅适用于一小部分蛋白质[117];
- 其次,从其蛋白质背景中分离构象B细胞表位以产生选择性抗体是一项困难的任务,需要合适的支架用于表位移植。 因此,构象B细胞预测目前与表位疫苗设计和基于抗体的技术无关。
尽管如此,预测构象B细胞表位对于进行涉及抗体 - 抗原相互作用,结构 - 功能研究是有意义的。
有几种可用的方法来预测构象B细胞表位(表2)。
- 第一个被引入的是CEP [118],它几乎完全依赖于预测溶剂暴露残留物的补丁(patches)。
- 其次是DiscoTope [119],除溶剂可及性外,还考虑氨基酸统计和空间信息来预测构象B细胞表位。使用59个构象表位的基准数据集对这两种方法进行独立评估,发现它们的精确度不超过40%,召回率为46%[120]。
- 随后,开发了更多的方法,如ElliPro [121],旨在识别抗原表面的突出区域,
- PEPITO [122]和SEPPA [123]它们结合了氨基酸的单一物理化学特性和几何结构特性。
这些方法的报告曲线下面积(AUC)约为0.7,这表明辨别能力差但优于随机。尽管如此,在独立评估中,SEPPA的AUC为0.62,而所有提到的方法的AUC均为0.5左右[124]。 ML还已用于预测3D结构中的构象B细胞表位。相关的例子包括EPITOPIA [125]和EPSVR [126],它们分别基于朴素贝叶斯分类器和支持向量回归,训练在组合不同分数的特征向量上。报告的这两种方法的AUC约为0.6。
上述构象B细胞表位预测的方法,无论抗体如何都能识别一般的抗原区域,这些区域被忽略[127]。然而,还存在抗体特异性表位预测的方法。这种方法由Soga等人开创。 [128]在分析抗原 - 抗体3D结构的界面后定义了抗体特异性表位倾向(antibody-specific epitope propensity,ASEP)指数。使用该指数,他们开发了一种新方法,用于预测单个抗体中的表位残基,其通过缩小常规方法预测的候选表位残基来起作用。最近,Krawczyk等人。 [129]开发了EpiPred,这种方法使用类似对接的方法来匹配抗体和抗原结构,从而鉴定抗原上的表位区域。 PEASE [130]使用类似的方法,并补充说该方法利用抗体的序列和抗原的3D结构。简而言之,对于每对抗体序列和抗原结构,PEASE使用机器学习模型训练来自120种抗体 - 抗原复合物的性质,以鉴定来自抗体的互补决定区(CDR)和与其接触的抗原的残基的对组合。
鉴定具有已知3D结构的蛋白质中构象B细胞表位的另一种方法,是通过基于模拟表位(mimotope-based)的方法。 模拟表位是选自随机肽文库的肽,因为它们能够结合针对天然抗原产生的抗体。 基于模拟表位的方法需要输入抗体亲和选择的肽和所选抗原的3D结构。 使用模拟表位进行构象B细胞表位预测的生物信息学工具的实例包括MIMOX [131],PEPITOPE [132],EPISEARCH [133],MIMOPRO [134]和PEPMAPPER [135](表2)。
如前所述,构象B细胞表位预测的方法通常需要抗原的3D结构。 然而,在例外情况下,Ansari和Raghava [136]开发了一种方法(CBTOPE),用于从抗原的一级序列中鉴定构象B细胞表位。 CBTOPE基于SVM并且训练构象B细胞表位的物理化学和序列衍生特征。 CBTOPE报告的交叉验证实验的准确率为86.6%。
四、结束语 Concluding Remarks
目前,T细胞表位预测比B细胞预测更先进和可靠。然而,虽然可以通过实验确认预测的大多数肽的MHC分子的预测结果,但只有约10%的那些被证明具有免疫原性(能够引发T细胞反应)[68]。如此低的T细胞表位发现率是由于我们还没有足够的模型来预测抗原加工[68]。低T细胞表位发现率的经济损失可以至少部分地通过优先化蛋白质抗原进行表位预测来克服[137-139]。对于T细胞表位疫苗开发,研究人员还可以求助于表位数据库中可用的实验已知的T细胞表位,通过免疫信息学选择提供最大种群保护覆盖率的那些[64,140,141]。在任何情况下,T细胞表位预测仍然是T细胞表位作图方法的组成部分。相反,B细胞表位预测效用目前更加有限。这有几个原因。
- 首先,对于线性和构象B细胞表位,B细胞表位的预测仍然是不可靠的。
- 其次,线性B细胞表位通常引发不与天然抗原交叉反应的抗体。
- 第三,绝大多数B细胞表位是构象的,但预测构象表位的应用很少,因为它们不能从它们的蛋白质背景中分离出来。
在这种情况下,关键不仅是改进B细胞表位预测的现有方法,而且还开发用于表位移植到能够替代天然抗原的合适支架上的新方法和平台。
总之,我们希望做两个与表位疫苗设计相关的最终评论。 首先,表位预测方法可以提供来自任何给定蛋白质查询的潜在表位,但并非所有抗原都与疫苗开发同等相关。 因此,研究人员还开发了用于鉴定疫苗候选抗原[142,143]的工具,这些抗原可能诱导保护性免疫,然后可以将其作为表位预测和表位疫苗设计的靶标。 其次,应该记住,表位肽表现出很小的免疫原性,需要与佐剂组合使用,佐剂通过诱导强大的先天免疫反应来增强免疫原性,从而实现适应性免疫[144-146]。 因此,新佐剂的发现与基于表位的疫苗特别相关[146],并且为此,Nagpal等人[147]开发了一种先驱的方法,可以预测RNA序列的免疫调节活性。
参考资料
- Journal of Immunology Research Volume 2017, Article ID 2680160, 14 pages. Fundamentals and Methods for T- and B-Cell Epitope Prediction ( 3.2分左右的杂志 ) https://www.hindawi.com/journals/jir/2017/2680160/
