【2】生物模态优化的计算方法
一、介绍
蛋白质及其复合物可以被认为是具有精细三维(3D)结构的分子机器,沿着在生物体生理学中有效的复杂生物途径执行特定任务。蛋白质的这种观点 - 它们的结构,相互作用和功能(their structure, interactions, and functio)-是大多数现代药物发现的基本要素。上个世纪生物和生物化学科学的发展促使药物开发活动从经验发现转向理性知识和数据驱动的方法。尽管如此,它被认为是一种偶然的发现,它仍然是药物发现的重要元素,并且是生物有机体复杂性的指标。跨越基因组学和分子生物学领域的工作与蛋白质的生物化学,晶体学和物理化学相结合,为推进蛋白质作用的机理观点及其在人类疾病中的作用提供了必要的知识基础。
随着我们对蛋白质及其行为的了解越来越多,结构-功能关系中的结构概念已经超出了X射线晶体学中通常观察到的范围,X射线晶体学是生成蛋白质详细结构洞察力的最强大技术。人们越来越认识到蛋白质是构象动态的实体。例如,蛋白质-配体识别, a central event in the mechanistic pathway,更好地被认为是涉及从相互作用蛋白质可用的几何体集合中捕获优选构象的事件,而不是埃米尔菲舍尔(Emil Fischer)在1894年提出的关联合作伙伴的严格锁钥契合。 由结合配偶体诱导的内在构象变化,以及与这种构象变化相关的动力学和自由能成本的方面,被认为是识别的关键组成部分。因此,沿着蛋白质活性的结构-功能轴,通过考虑结构,动态和能量方面来最好地理解该系统。构象动力学导致蛋白质作为构象子基团在自由能源景观上的集合,其具有障碍和它们之间的动态转变。表征蛋白质固有的构象多样性和构象子位之间的平衡,对于鉴定有助于蛋白质稳定性和结合性的因子是重要的。在这方面,计算机蛋白质建模和模拟将计算算法与经验数据,序列和结构信息相结合,以开发对系统及其动态的详细和现实的评价。这使人们能够形成批判性和新颖的物理化学见解,扩展到动态结构-功能关系的范式。分子建模和计算模拟在研究蛋白质及其复合物的这些动态和热力学方面具有独特的优势。 2013年诺贝尔奖授予Martin Karplus,Michael Levitt和Arieh Warshel在这一领域的工作,强调了计算生物分子建模和模拟的价值。随着药物发现和开发领域从小分子领域扩展到蛋白质治疗领域,计算机rational蛋白质工程也在扩大其在生物开发中的作用,作为一种关键工具,可以补充其他实验探索手段。
使用合理的蛋白质工程方法可以有效地优化治疗蛋白质的方式,解决一系列问题,包括稳定性,有效性,药代动力学,药效学和表达生产力。 虽然传统的设计质量(quality by design, QbD)工作侧重于过程控制和优化治疗的发展以实现产品质量,但有人呼吁将QbD扩展到整个药物开发过程。结构指导方针对治疗性蛋白质的工程设计可以提供有价值的科学洞察力,旨在降低其发展的风险,这是QbD核心的理念。 本章将回顾计算方法在药物发现和开发的合理结构和知识驱动方法中的应用,重点是设计基于蛋白质的治疗作用和活性的方式。
二、蛋白质治疗优化的计算技术 ( COMPUTATIONAL TECHNOLOGIES FOR PROTEIN THERAPEUTIC OPTIMIZATION )
虽然计算机辅助药物设计领域在小分子药物(small-molecule drug, SMD)发现中具有非常明确的作用,但计算生物设计领域正在发展以确立其地位。图2.1展示了有效使用计算平台进行合理结构引导生物工程所必需的各种技术,目的是预测实现感兴趣的生物物理或功能特性的突变。准确地模拟蛋白质及其变体类似物,捕获其构象灵活性的各个方面以及与其他蛋白质和分子伙伴的相互作用,了解控制和定义这些特性的能量标准,并开发软件和硬件以使这种方法易于处理多学科的努力。图中的组件被表示为拼图,以强调这样一个事实,即尽管可能会针对每个部分进行广泛而详细的工作,但将多样化的部分组合到一个集成的技术平台中具有挑战性。

图2.1 用于蛋白质治疗剂的分子优化的计算平台的组件。 本图介绍了Zymeworks解决的分子模拟领域,以开发可用于合理结构和计算模型引导的候选蛋白质治疗工程的工具链。 生物优化计算平台的开发和应用是一项多学科的研究,需要在化学,物理,结构生物学,数学,数据挖掘和计算机科学领域的专业知识。
2.1 模型构建与结构分析
用于准备3D结构和执行模型或模拟输出分析的分子建模基础架构,是该技术的核心部分。建模(Modeling)和模拟(simulation)非常容易受到“垃圾进入,垃圾输出(garbage in, garbage out)”这一格言的影响,在这方面,结构准备解决了典型的结晶学衍生结构的假象,并读取了建模的质控数据。分配质子化(protonation)状态,特别是对于显示pH依赖性活性的系统,是模型质量的重要因素。识别和明确模拟蛋白质表面或内部的关键水分子和离子的能力是影响模型质量的另一个因素。文献中有丰富的例子,其中蛋白质已被证明采用结合水分子作为氨基酸的延伸酸侧链实现特定的功能。获得新蛋白质结构的比较建模是该工具链中的关键技术,将在本章后面的抗体结构建模的背景下更详细地解决。
基于分子建模框架(molecular modeling infrastructure)的结构分析,可以揭示许多可能影响蛋白质特性的相关特征,并有助于发现和优化步骤的理解。图2.2显示了蛋白质结构中的特征列表,这些特征有助于指导结构 - 功能关系理解。蛋白质结构分析的各个方法的细节可以在其他地方找到。基于这种数据丰富的分析的机器学习方法可用于绘制关于蛋白质结构 - 功能关系的新颖见解。基于这种详细的结构分析,个体或残基组的重要性可用于计算蛋白质工程中可能有利或有害的热点或蛋白质中的位置。高质量的高分辨率晶体结构和新颖的室温X射线晶体学的使用提供了直接证据,即结构中的原子可以占据多个位置,从而呈现多种交替的构象状态。有一些主要信息表明这些残基构象状态之间的变构相关性能够在蛋白质结构内形成功能相关的残基网络。理论分子模拟及其应用的发展,能够提供关于蛋白质结构动力学及其对功能活动的贡献的复杂细节。
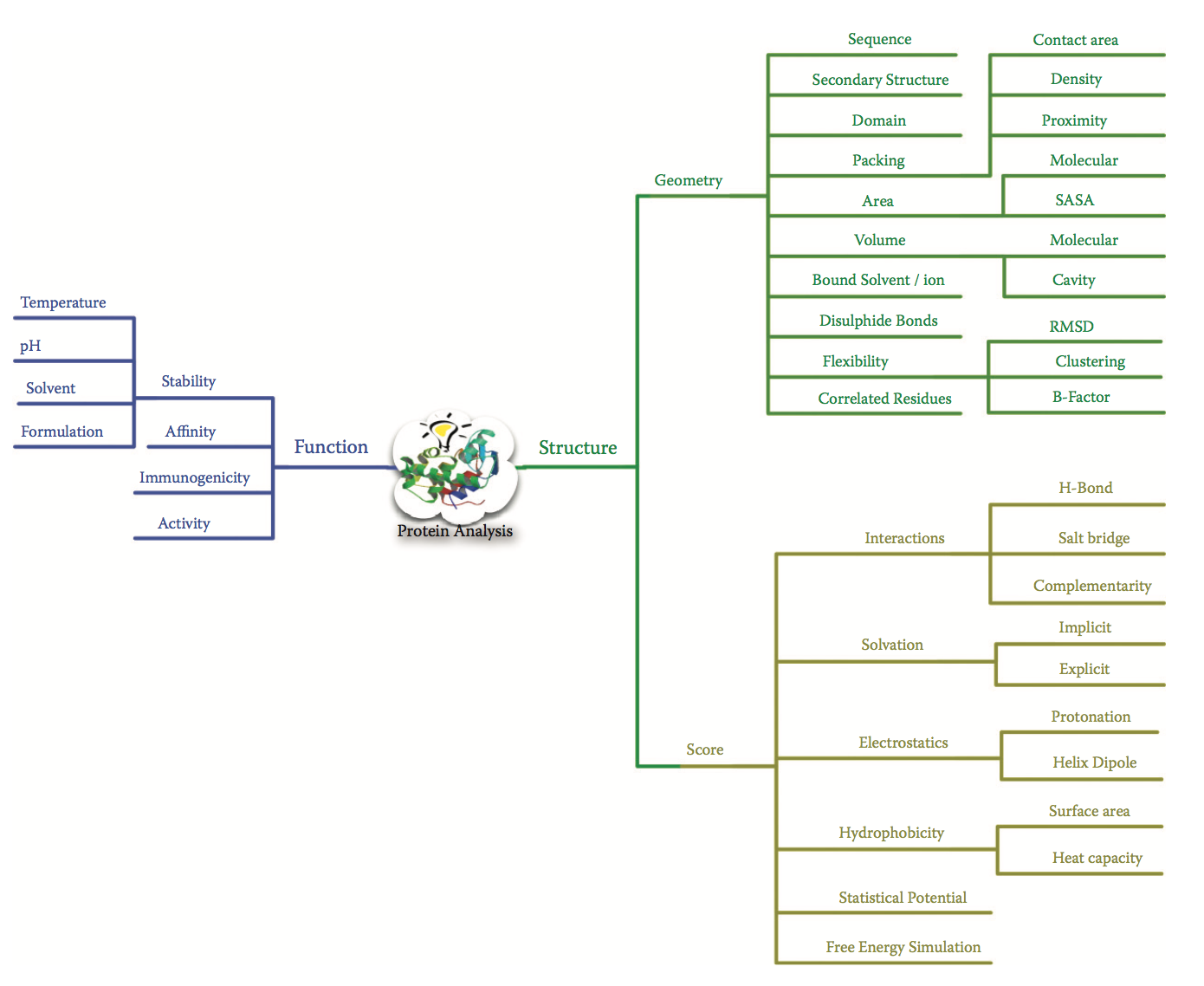
图2.2 与蛋白质结构和模型分析相关的问题和方法的“思维导图”。 虽然蛋白质优化通常针对稳定性或物质改进,但该方法将涉及深入研究结构或几何方面,以及有关其能量方面的信息。
2.2 构造变化与动力学的模拟
对蛋白质进行建模以及与引入的突变相关的结构变化,需要使用适当的取样方法来了解蛋白质及其突变体所采用的潜在构象。使用模拟来模拟蛋白质构象变化有助于扩展通过诸如晶体学的方法得到的结构信息的效用。增加的信息是关于系统处于改变状态的见解,受蛋白质的外在或内在条件的影响。从算法的角度来看,蛋白质构象抽样方法可以被分类为确定性(deterministic),随机(stochastic)或组合确定性和随机的方法。确定性模拟涉及以可预测的方式演化系统的算法。在分子动力学等确定性方法中,系统中的原子或粒子是根据牛顿运动定律模拟的,粒子运动是由与其系统中的其他粒子相互作用而作用于它的力决定的。在短时间步骤期间迭代地计算这样的有效力和合成位移,以便预测系统构象变化的时间演变。虽然能够提供有关蛋白质动力学的详细信息,但分子动力学可以从启发式方法中获益,这些方法可以指导其他方法的相关感兴趣领域的构象抽样。诸如蒙特卡罗模拟的随机方法在构象空间中执行随机移动,并且使用诸如Metropolis Monte Carlo标准的方法可以使这种采样更有效,所述方法接受或拒绝具有某些基于能量的概率条件的构象变化。这种基于蒙特卡罗(Monte Carlo)的构象搜索方法,可以适应于基于蛋白质结构内的各种自由度来对蛋白质运动进行采样。
蛋白质构象采样方法解决局部几何特征的不同方面,如氨基酸侧链拓扑,骨骼几何和键向量方向变化,循环几何描述,二级结构元素的重组,域-域接触的优化,鉴于时间尺度的不同,人们可能期望这些变化,并且蛋白质-溶剂表征已被解决并作为独立问题解决。在这些方法中,已经证明基于旋转异构体的氨基酸侧链构象取样方法的各种方法,在蛋白质工程应用的背景下特别有价值。这些方法重新优化了局部区域的蛋白质包装,并且非常有效地计算筛选不同氨基酸类型在计算机优化的背景下感兴趣的位置的影响。必须采用各种这样的近似构象采样方法,因为允许使用诸如分子动力学模拟之类的技术对蛋白质的完整构象空间进行显式和不受限制的采样在实际时间表中仍然是计算上难以处理的,特别是以高通量方式。数十万种突变可以在蛋白质工程中筛选出来。随着越来越多的努力采用高度专业化的高性能计算硬件和图形处理单元(GPU)来解决这种迭代数值问题,这种情况正在慢慢改变。由于局部相互作用,可行性和折叠倾向的改变,蛋白质结构,突变的性质及其对稳定性的影响之间存在着复杂的联系。蛋白质构象建模和分子动力学模拟提供了有价值的工具来评估这些问题,分析和发展了有关蛋白质及其突变的结构方面的见解。
2.3 能量函数和打分
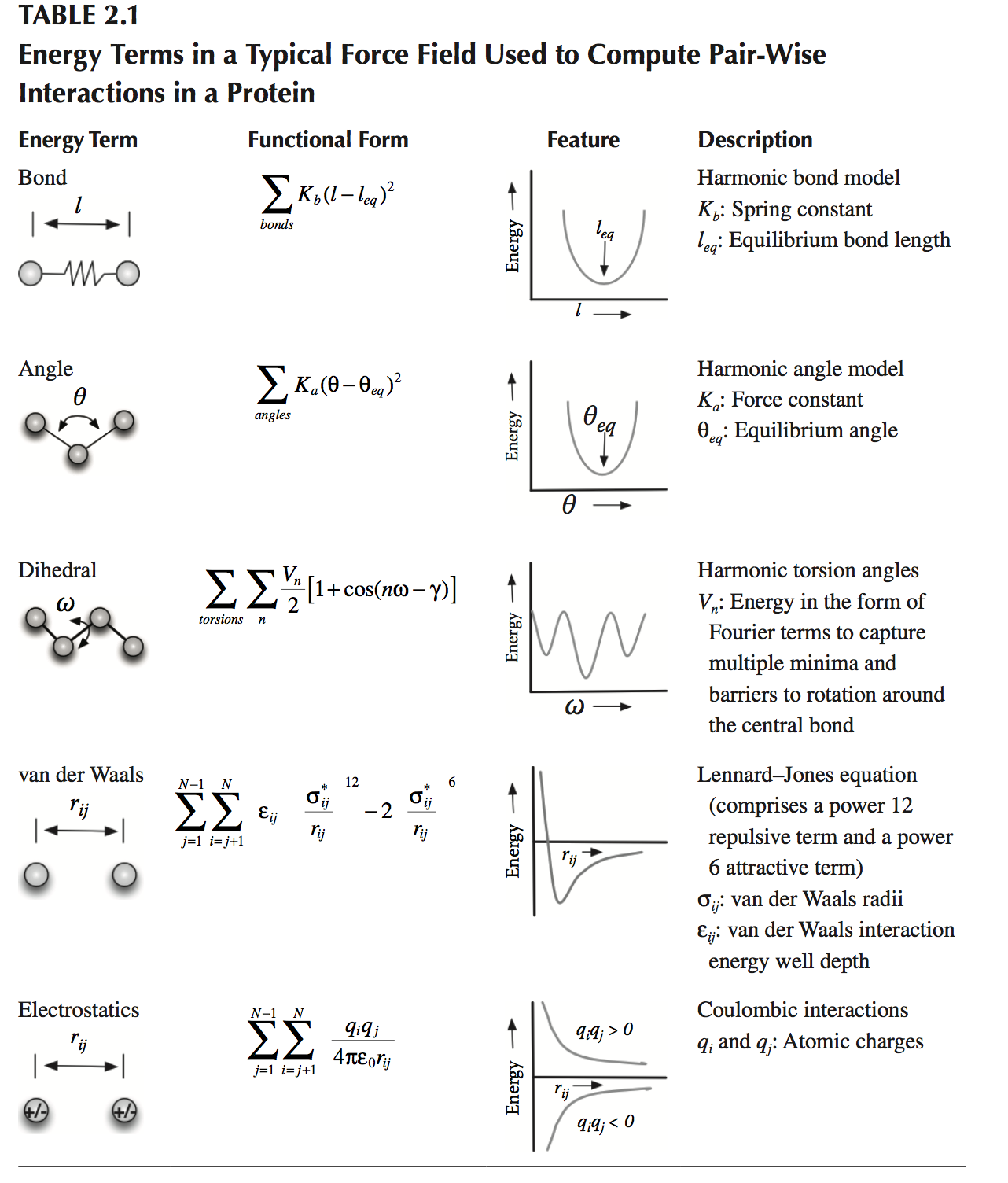
决定蛋白质模拟可靠性的另一个关键因素是能量函数的性质,它模拟蛋白质和环境中颗粒的相互作用。蛋白质模拟的典型能量函数基于从高水平从头量子力学计算得到的有效对势,以及从分子分析得到的数据,如光谱学,晶体学和作为氨基的小分子的溶剂化能量酸类似物。用于蛋白质模拟的基于物理的势能函数(通常称为力场,force eld)通常包括捕获蛋白质的拓扑特征的功能术语,例如键(bonds),键角(bond angles)和扭转角( torsional angles)。与键长和角度变化相关的能量被描述为谐波函数(harmonic function)。扭转项表示为有限傅立叶展开(Fourier expansion),以描述关于键的旋转的立体化学效应。此外,成对的非键合相互作用项包括模拟普遍但弱的范德华(分散 - 排斥)相互作用的函数,通常由Lennard-Jones方程建模,以及库仑(Coulomb)方程模拟的电荷 - 电荷(静电)相互作用。
表2.1列出了这些能量项的更多细节和功能形式。范德华相互作用具有短程,这表明可以通过切断(即避免)超过某个原子间距离的相互作用来近似它。另一方面,静电效应的范围更长。
作为对蛋白质环境中众多溶剂分子(水和离子)的明确处理的替代方案,这对于计算要求很高,溶剂化的热力学效应 可以在代表溶剂的介电屏蔽效应的静电的连续模型近似的基础上相当准确地建模。在这方面,Poisson-Boltzmann处理溶剂环境的方法对于估计溶剂化和结合的静电效应特别有价值。疏水效应的处理,与蛋白质表面非极性组分周围溶剂重排的熵效应有关,通常根据蛋白质溶剂可及表面积的变化来模拟。
单个类似物或突变体相对于彼此或与亲本蛋白质的准确评分和排序是计算机辅助方法中最具挑战性的方面之一。从正确的角度来看,折叠蛋白质的稳定性通常比参考未折叠状态的稳定性大约5-10千卡/摩尔,而良好形成的相互作用(如一个氢键)的强度可能与结果,即使在这些关键相互作用的描述中存在很小的不准确性,也会使模拟结果在数量上不正确。
已经开发出严格的自由能和预测系统状态之间的热力学自由能差异的平均力模拟方法的潜力,但是能量函数中的固有近似和在这些模拟中实现收敛的计算成本使得它们经常使用极具挑战性。来自高分辨率3D蛋白质结构数据库的残留物或原子配对概率已被用作校正和发展与蛋白质相关的统计潜力(也称为基于知识的潜力)的手段。这种基于知识的潜力本质上代表所有热力学因素导致蛋白质折叠状态的影响。
鉴于这些计算算法试图仿效分子结构背后极其复杂的物理学,以及与相当简单的有效对电位的相互作用这一事实,有很多机会重新定义和优化这些方法,以便将它们用于预测方式。 从计算方法中获取价值的一种有效方法,记住它们的局限性,是通过参与计算和结构引导建模的设计周期来提出结构 - 功能假设,然后对假设进行实验评估。 具有实验数据的迭代反馈和模型训练有助于为后续优化提供假设
三、计算方法在蛋白质治疗优化中的应用
3.1 目标(TARGET)发现和计算方法
药物发现的合理方法的核心,是对作为治疗目标疾病唯一相关的生物分子的鉴定和广泛表征。 靶标通常是在疾病状态下表现出改变的表达的蛋白质,通常具有促进疾病进展的功能特征。 它可能涉及属于受体,酶,离子通道,生长因子,细胞因子,核酸结合蛋白或与这些信号元件相互作用的其他因子类别的信号转导途径。 候选药物通常结合和/或调节靶标的活性以影响疾病状况。
导致制药工业生物制剂显着增长的众多因素之一,是可以用小分子药物(small-molecule drugs,SMD)有效治疗的蛋白质靶标数量的限制,即成药性(druggable)。 基因组和蛋白质结构生物信息学在获得基因组中可药用蛋白质空间的高水平图像,以进行目标识别方面发挥着主导作用。 已经指出,有限的SMD效用起源于靶标中结构元件的不充分分化,允许小药物样分子以非常高的亲和力和特异性(药物可能性)结合所必需的分化(differentiation)。 生物制剂,特别是抗体,可以有效地解决与特定靶向相关的这些问题。 最近有关临床试验阶段转换的统计数据表明,生物制剂批准的可能性几乎是SMD的两倍。
在目标识别的背景下,人们越来越认识到复杂生物系统的现实不利于还原单一疾病,单一目标,单一药物模式。尽管这些机制模型在药物开发中具有巨大的实用性,但人们不能忘记药物作用的生物学通过其对涉及多种蛋白质共同作用的信号级联的影响而在系统水平上发生。药物对其结合的靶标之外的元素有显着影响,因此靶标是生化系统而不仅仅是蛋白质。这引入了复杂的毒性问题,使得目标验证的转化方面极具挑战性。这也为数据和信息驱动的药物发现和优化方法提供了机会。对目标验证的强调引起了人们对开发生物标记和最佳生物制剂的浓厚兴趣,这一领域得到了理性药物发现和优化工作的良好服务。 Biobetters,也称为biosupeiors,是潜在的下一代疗法,包括针对已知目标的已知生物治疗剂的版本。生物标准物(Biobetters)通常被设计用于改善功能或物理化学性质,并且对效果具有显着影响。通过计算优化服务的大量治疗剂属于生物标记物的类别。
系统生物学是生物信息学的一种增长途径,其应用于目标的识别和新型生物制剂的开发。细胞环境中活跃的生物化学网络中的众多分子参与的系统方法,可以帮助更深入地理解诸如癌症的复杂疾病。对这种生物网络进行建模依赖于从各种高通量“组学”技术得到的经验数据,这些技术与这些动态网络的复杂数学和统计模型相结合。对这些模型的分析有助于产生对生物系统的新的预测性见解以及将这些知识应用于药物发现的机会。 Schoeberl及其同事使用ErbB信号通路的计算模型来捕获该系统中各种已知配体和抑制剂的活性,并使用该信息来鉴定治疗候选物中感兴趣的靶标和特征。系统生物学可以在个体化医学的发展和由致病细胞进化的抗性机制的鉴赏中发挥重要作用。
3.2 生物活动的方式
生物制剂的使用可以根据其作用方式大致分为三个治疗组。第一个治疗组由外源或重组衍生的酶或调节蛋白组成,用于治疗患有内源性或内源性蛋白异常的患者。重组细胞因子或生长因子可用于增加某些疾病状况下的天然活性。第二类涉及使用抗体或其他特异性靶结合蛋白,其可能干扰靶蛋白的活性或其参与或介导其他化合物或蛋白的递送的途径。抗体是一种非常通用的分子,涉及许多功能相关的分子相互作用,从治疗设计的角度来看,这可能很重要(图2.3)。工程化双特异性抗体的新兴领域代表了靶向结合蛋白类别下的新类别,其中药物能够同时结合两种不同的靶标以实现其活性。在随后的章节中,我们将回顾使用计算机蛋白质工程技术来优化这两大类生物制剂中的治疗方法。第三类生物制剂由蛋白质疫苗组成。虽然有许多已建立的计算机技术支持第三类生物制剂,例如疫苗设计,T细胞表位的鉴定等,但本章未涉及这一途径。
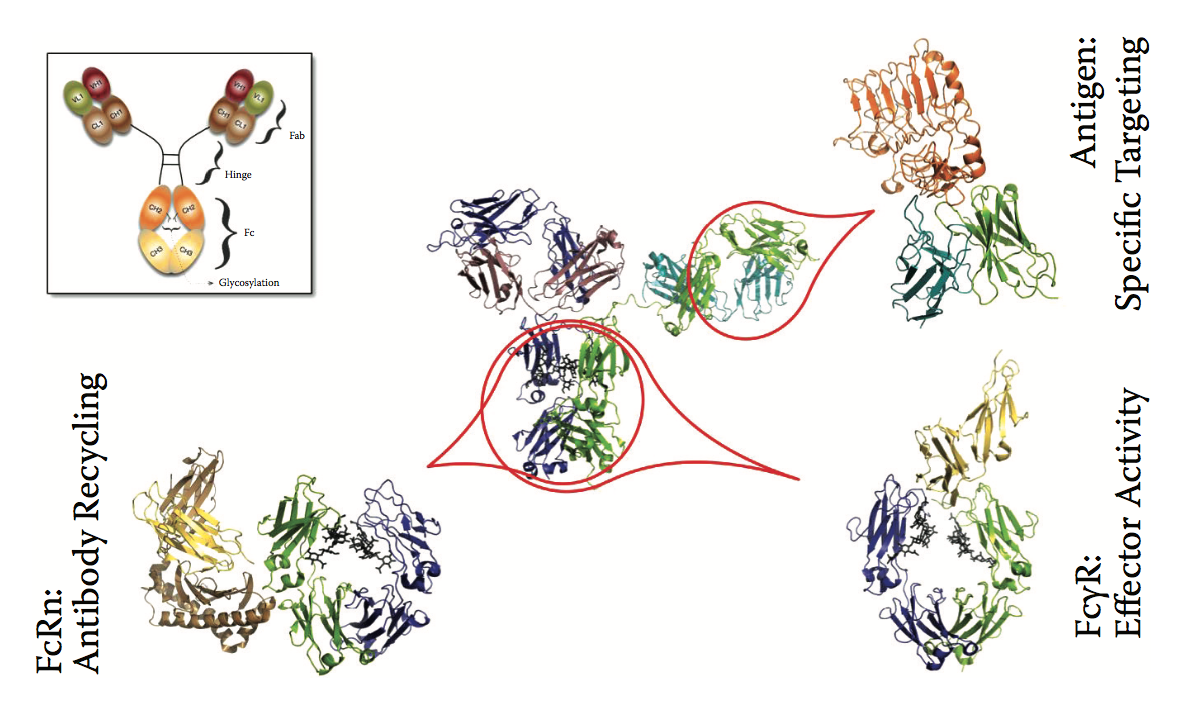
图2.3 抗体是一种通用的多功能大分子,能够进行涉及其不同区域的各种功能重要的蛋白质 - 蛋白质相互作用。 该图提供了具有各种其他蛋白质的抗体结构域(亚基)的代表性结构,特别是Fc结构域与FcRn和FcγR蛋白质的复合物,以及涉及通过可变结构域的末端CDR处的CDR的相互作用的代表性抗原-Fab复合物结构。 (蛋白质数据库[PDB] ID:1HZH,1A1X,1E4K)。 在原子级别上这种详细结构信息的可用性为这些交互的结构引导优化提供了机会。 插图图示了抗体的示意图( schematic representation)。
3.3 胰岛素的优化
调节蛋白治疗剂的最突出的例子是胰岛素。用于糖尿病管理的胰岛素和胰岛素类似物的优化和开发,证明了合理工程化生物疗法可用于实现更好的作用方式的机会。胰岛素参与与葡萄糖代谢和控制(体内平衡)相关的功能,在不同类型的糖尿病患者中不同程度地存在。人体中的活性胰岛素是翻译后修饰的蛋白质,其包含两条链(A和B),分别长21和30个氨基酸,通过三个半胱氨酸二键交联。胰岛素的晶体结构是通过X射线衍射解决的第一种蛋白质。
胰岛素可以作为溶液中的单体(monomer),二聚体(dimer)或六聚体(hexamer)存在。六聚体构型包括结合的Zn2+离子,并且在胰腺细胞中起胰岛素储存的作用,而解离形式具有生物活性。为了在治疗环境中概括胰岛素分泌的生理模式并实现葡萄糖稳态,已经开发了各种蛋白质工程改造的胰岛素类似物,改变其治疗方式,特别是其药代动力学特性。关于胰岛素的可用结构信息具有帮助工程工作,针对二聚体和六聚体形式的界面形成中涉及的残留物。使界面不稳定已经产生速效胰岛素,同时稳定界面产生在血清中以二聚体或六聚体存在的制剂,产生缓释药理学特征(表2.2)。六种不同的胰岛素类似物的批准以及定制的药理学,描述了使用蛋白质疗法的结构功能理解可以实现的目标。最近的努力旨在通过设计新的锌结合配置来设计延长释放形式,所述新的锌结合配体能够形成具有潜在更好性质的胰岛素六聚体的超分子组装体,例如延长释放和储存稳定性。

3.4 药代动力学优化 PHARMACOKINETIC OPTIMIZATION
虽然小分子药物通常具有口服递送以通过胃内层吸收进入血液的优势,但功能活跃的蛋白质治疗剂在摄入循环系统之前不能在富含蛋白酶的口服/胃途径中存活。蛋白质治疗剂通常通过肠胃外途径(皮下或静脉内注射)递送,因此,频繁给药不是维持必需血清浓度的非常方便的选择。已经致力于开发合理的蛋白质工程方法,其可以改善治疗性蛋白质的药代动力学(PK)性质,从而提高其生物利用度和使用方便性。一旦进入血清,蛋白质治疗剂的命运就是其大小,以及物理和化学性质(例如电荷,疏水性和形状)的函数。肾脏对小于60kDa的蛋白质的过滤,受体介导的清除以及随着抗药物抗体的发展的免疫原性应答是从循环系统中去除的显着途径。
许多小的治疗相关蛋白,包括胰岛素,人生长激素(rHGH),集落刺激因子(CSF),促红细胞生成素(EPO),干扰素α-2a和α-2b,以及人白细胞介素(IL-11) ,当用作治疗剂时,已经显示出具有短的血浆半衰期。增加治疗性蛋白质的大小和流体动力学半径提供了合理地改善血清半衰期的手段。在这方面实现蛋白质治疗剂的生物质标记形式的方法之一是将蛋白质与惰性聚合物(例如聚乙二醇(PEG))缀合。聚乙二醇化版本的重组G-CSF和干扰素α-2a和α-2b以及腺苷脱氨酶被批准用于临床,并显示出改善的PK特性。这种结合对靶标结合和药效学活性的潜在影响是这种努力中明显的后续关注。可以采用结构指导的方法来设计PEG化位点而不会不利地影响治疗效果。
在EPO的情况下,Amgen的科学家开发了一种糖基化改造版本,其包含5个氨基酸取代,通过引入共有位点NXS / T序列引入两个新的N-糖基化位点,此外还有三个N-糖基化和存在于亲本重组蛋白中的位点一个O-糖基化。当重组产生时,这些取代导致具有5个碳水化合物链和多达22个唾液酸残基的EPO形式。由于唾液酸在分子上添加额外的碳水化合物链和负电荷,分子的血清半衰期几乎提高了三倍,因此表现出更好的效果。在进行这项工作时,没有详细的结构信息,因此研究人员用共识位点筛选了数十种类似物,以识别两个功能活跃的位置,这些位置随后以相同的顺序组合以实现感兴趣的类似物。有两个新的碳水化合物链。从结构角度来看,糖基化位点的引入将需要选择引入的碳水化合物部分将保持溶剂暴露并且不显着影响局部蛋白质构象的位置。已经尝试了结构引导的方法,以有效地引入新的糖基化位点,其具有所需的其他治疗候选物的血清半衰期的改善。
3.5 FCRN受体和生物学
设计治疗性蛋白质候选物与抗体Fc的融合是另一种改善其血清半衰期的策略。或者,与人血清白蛋白,或与白蛋白结合的肽或其他小分子的融合可以增强治疗候选物的血清半衰期。抗体Fc和白蛋白以pH依赖性方式与内皮表面上的FcRn受体相互作用,在酸性pH下结合并在生理或中性pH下释放。在此过程中,抗体Fc和白蛋白在胞饮作用后逃避溶酶体降解,这是血清中大多数蛋白质的命运。结构分析结合实验筛选已被用于指导这种相互作用的结构 - 功能模型的发展,进一步优化这种迷人的救援机制。 这些分子事件中有一些非常微妙,对整体效应进行建模可能具有挑战性,要求在构象变化的模拟和质子化状态和结合的耦合变化方面具有高精度。最近的一项研究表明,功能活跃的突变可能与Fc-FcRn界面相距很远。这些信息正在指导具有改变的药代动力学特性的变体的设计和工程,目的是可能使用它们开发具有更长血清持久性的生物上层版本的重磅抗体。另一方面,在一些治疗设计中,可以设计氨基酸突变以操纵FcRn结合,使得治疗或治疗靶标复合物优先分选用于降解
3.6 FCγ受体(FCγR)亲和力调节
鉴于Fc在抗体和Fc融合分子中的广泛用途,已经花费了大量努力来设计Fc以用于定制的功能活性。特别地,抗体结合靶(细胞)并通过Fc募集免疫效应细胞以介导靶杀伤的能力,是基于抗体的治疗作用的重要方式。存在于免疫细胞上的FcγR可以与免疫复合物中的抗体Fc结合,以诱导针对靶细胞的效应子活性,例如抗体依赖性细胞毒性( antibody-dependent cellular cytotoxicity ,ADCC)或抗体依赖性细胞吞噬作用( antibody-dependent cellular phagocytosis,ADCP)。类似地,Fc与补体系统的C1q蛋白的相互作用可以诱导补体依赖性细胞毒性(complement-dependent cytotoxicity,CDC)活性。免疫效应细胞在其细胞表面上表达多种FcγR,其在Fc结构域识别的细胞外区域中特别同源。这些受体在它们所含的细胞内免疫受体酪氨酸激酶基序上不同,它可以起到激活(ITAM)或抑制(ITIM)信号的作用。在人类中,FcγRI(CD64),FcγRIIa(CD32a)和FcγRIIIa(CD16a)含有ITAM基序并诱导活化免疫应答,而FcγRIIb(CD32b)具有能够负调节B细胞免疫应答的ITIM基序。另一类Fcγ受体FcγRIIIb缺乏酪氨酸激酶基序,而是具有糖基磷脂酰肌醇(GPI)锚。根据与疾病状况相关的免疫细胞类型及其Fcγ受体表达水平,以及每种这些受体的Fc相对亲和力,可以调节免疫细胞的参与程度及其反应。已经进行了许多结构指导的抗体工程努力,目的是实现调节免疫效应子活性的Fc类似物。 工程化Fc以选择性结合FcγRIIIa或FcγRIIa已经产生具有增加的活化剂/细胞杀伤活性的抗体类似物,而FcγRIIb已经被靶向以实现抑制性应答。鉴于广泛使用Fc融合策略以主要改善治疗候选物的PK特性,还采用结构指导策略来改造突变以敲除由Fc-FcγR相互作用诱导的天然效应子活性。
3.7 FC融合和二聚化
除了由含包含Fc分子产生的基于FcRn和FcγR的活性之外,还开发了作为Fc融合体的治疗性蛋白质或肽的工程化,以利用Fc的同源二聚化特征和这些融合可以提供的一系列的拮抗或激活活动。 已经显示,在abatacept的情况下,CTLA-4与Fc的融合,作为二聚体的交叉臂结合允许更强烈地结合其靶标CD80和CD86。 Belatacept是一种合理工程化的abatacept变种,具有两个额外的突变,比CD86更加紧密地结合四倍,并且对CD80更加狂热,其效力增加了十倍。
3.8 双特异性抗体 (BISPECIFIC ANTIBODIES)
除了其高度特异性的靶识别特征外,抗体与多种靶/受体蛋白同时相互作用的能力对于其作为相对于SMD的新型治疗剂的实用性至关重要。作为一个例子,利妥昔单抗(rituximab)通过其Fab结构域与B细胞抗原CD20相互作用促进免疫复合物的形成,而Fc结构域与免疫效应细胞相互作用,从而桥接细胞并促进抗体依赖性细胞毒性的响应。用能够介导两个或多个靶标或表位之间接触的多特异性蛋白质治疗剂实现新的作用方式的想法,已经成为开发被称为双特异性(bispecifics)的工程化治疗形式的驱动力。
双特异性分子(bispecific molecular)格式或支架的设计已经成为蛋白质工程努力的方法,经过不同程度的评估zoo of molecular formats。将这些不同的分子形式转化为真正的治疗候选物质,会对工程解决方案的可开发性提出问题。 。从分子平台或支架的角度来看,可开发性是指支架在不同系统中以即插即用的方式易于使用,以便针对感兴趣的目标测试和筛选双特异性活性。治疗候选物的观点,超越了分子的PK和药效学(PD)形式的有效性,可开发性解决了产品开发的各种其他CMC方面,如可制造性,质量控制和商品成本,以实现产品均匀,可重复,易于分析,易于配制,并且在必要的药物浓度下稳定。在开发初期对蛋白质的基本生物物理特性进行详细评估,在初始化过程开发之前就此提供了大量见解。
3.9 异二聚体FC工程 ( HETERODIMERIC FC ENGINEERING )
通过工程化获得异二聚体(heterodimeric)Fc作为双特异性抗体框架(scaffold)的基础,可以改变Fc结构域同源二聚化(homodimerize)的固有趋势。这种设计的主要优点是它可以保留天然的三级结构和Fc其他相互作用倾向。 Carter,Presta及其同事在Fc的CH3界面采用的旋钮进入(knobs-into-holes,KiH)策略涉及通过用较大的一个(例如,T366W)突变较小的残基而在一条链上引入旋钮突变,在另一条链的互补表面上创建了一个容纳旋钮的孔,具有更大到更小的残留突变(例如,T366S,L368A和Y407V)。这种设计使系统偏向于优先形成异二聚体物种。然而,该设计导致CH3结构域相对于野生型Fc的显着稳定性丧失(~15℃),其随后通过噬菌体展示进行优化。
与KiH设计中的空间互补方法相反,Gunasekaran及其同事采用结构引导的静电荷反转(charge inversion,CI)设计策略来实现Fc的选择性异二聚化。基于序列和结构指导的工程方法,Davis及其同事设计了一个链交换工程结构域(strand exchange engineered domain, SEED)CH3,它由人IgA和IgG CH3序列的交替片段组成,产生优先结合的互补异二聚体。根据KiH设计的趋势,源自CI和SEED体方法的异二聚体CH3结构域导致~68°C的热稳定性。开发治疗剂以解决这种基本的生物物理稳定性损失是有利的。已经采用迭代结构和计算模型引导的蛋白质工程结合生物物理表征来设计Fc中的突变,其允许两个设计的互补Fc链的精确配对特异性。同时保留天然抗体样稳定性和可开发性属性。高通量生物物理表征和筛选技术的改进可以简化这种合理的优化方法。
3.10 抗体可变域建模
噬菌体和其他显示技术方法是最广泛使用的技术,用于在抗体开发的治疗应用的背景下解决抗原结合特异性和亲和力的成熟度要求。计算机辅助抗体设计方法刚刚开始在这方面取得进展,展示有限数量的成功案例。传统上,已经投入大量精力来使用同源信息来模拟抗体结构,其细节被用于模拟变体来改善相关特征,例如亲和力和稳定性。早期,结构建模是抗体(Fab)人源化过程的核心。该过程受到以下理解的指导:互补决定区(CDR)的序列和几何结构以及框架中有限数量的锚定残基对抗原结合是关键的。这种小鼠来源的人源化抗体构成占有被许可用于人类临床的抗体的很大一部分比例。
基于序列和结构指导的抗体Fab结构同源性建模,在抗体结构建模的领域被广泛使用。对CDR的几何形状进行建模,以及恒定和可变结构域之间的角度,是准确预测抗体结构的重大挑战。在与抗原构成基本超变量接触空间的六个CDR环中,对重链(H3)中的第三个环建模,其长度,序列和构象可以相当不同,通常对抗原结合贡献最大。并且是抗体Fab结构建模中最具挑战性的方面之一。可以基于序列同源性和其他已知抗体结构中的规范几何结构有效地模拟其余环的几何形状,其往往更短。使用固定的能量函数和定制的Ramachandran图作为指导构象采样的知识库正在推动这种H3循环建模工作的改进。 H3环几何的元素也与Fab中VL / VH结构域的方向偶联,这是抗体结构预测中的另一个关键变量。基于生物信息学的抗体结构元件鉴定和合成抗体文库的构建在设计中也是至关重要的。
在对抗体-抗原复合物进行建模时,它们的对接配置(docking configuration)和结合能力(binding affinity)是一个更大的挑战。与典型SMD的蛋白质 - 配体结合时埋藏的约400Å表面积相反,抗体-抗原界面通常埋藏约1200-2000Å2。虽然在复杂形成时获得的这种大的表面区域使得抗体赋予了精致的特异性,但是它使得结构引导的抗体工程的界面的精确建模非常复杂。构成界面的环的灵活性质及其熵贡献增加了精确模拟这些复合物中结合和亲和力的挑战。
人们越来越认识到远端变构效应( distal allosteric effects )可能在抗体结构及其作用中发挥作用。计算方法可用于进一步了解这些属性。当我们认识并开发使用抗体作为治疗剂的新方法时,定制的抗原结合特征对于合理地修改这些治疗剂的功能活性将是至关重要的。结构引导的诱变残基选择,可以通过实验亲和力成熟技术来加速最佳变体的开发。比较模型还可以帮助更好地理解抗体-抗原结合背景下的“序列-结构-功能”关系。在该领域的早期发展过程中,发表了一种结构引导方法,用于重新设计人源化抗体以实现物种交叉反应,从而促进临床前药物开发过程中的非人灵长类动物研究
3.11 药物输送系统 DRUG DELIVERY SYSTEM
人们对保罗·埃利希(Paul Ehrlich)治疗性神奇子弹的看法产生了极大的兴趣,该治疗性神奇子弹非常具体地将疾病治愈性毒素传递给靶细胞。与具有全身作用的非靶向细胞毒性剂相比,预期这些药剂具有更好的安全性,从而改善治疗的治疗指数。多年来,具有高度特异性靶向能力的抗体被认为是将共轭药物分子传递给目标疾病细胞的特洛伊木马,并且随着最近批准的brentuximab vedotin和ado-trastuzumab emtanine,该领域显示出恢复活力的兴趣。 。在这两种治疗方法的情况下,抗体-药物偶联物( antibody–drug conjugates,ADC)分别显示出比其亲本抗体SGN-30和曲妥珠单抗(trastuzumab)更好的活性。共轭分子(conjugated molecules)能够扩展治疗作用的方式,超过亲本抗体所实现的方式。 SGN-30已经在II期临床试验中进行了测试,其效果不大,而曲妥珠单抗治疗虽然具有革命性,但其原发性和治疗突发性耐药率很高,阻碍了长期预后。目标是通过ADC的工程和设计来管理毒性挑战,将有毒有效负载与高特异性靶向剂(如抗体)结合,可能会开启抗体和本身并没有显示出很大的希望的小药物样分子的效用,。
抗体靶向的受体,偶联(conjugation)中使用的药物,用于将药物与抗体结合的接头,以及抗体上偶联位点的位置都是决定ADC的PK,PD和毒性的重要因素。 在偶联的药物分子数量方面获得同质产物并保持结合抗体的稳定生物物理特性,与治疗窗口相关的特性是ADC发展挑战的关键因素。许多毒素分子例如,在ADC中使用的微管蛋白结合剂和DNA嵌入剂本质上是高度疏水的,需要认真考虑这些可能对偶联抗体产生的去稳定作用和产生偶联材料的复杂性。基于结构的建模已被用于识别可能有利于半胱氨酸突变的位置,随后用于在称为Thio-Man的抗体中结合毒素分子。在该研究中,显示偶联位点处的局部构象和生物物理性质影响药物释放和ADC的总体活性。已经尝试对这些Thio-Mabs进行分子动力学研究,以了解突变的影响和小分子与局部构象动力学的结合。认识到工程突变的位点,其中药物分子的缀合可能是有利的,需要详细的结构 - 功能理解药物缀合位点和药物缀合和释放的化学。在新蛋白质工程策略的背景下进行建模和模拟研究以了解这些细节将特别有价值,该策略旨在在蛋白质链中引入非标准氨基酸以实现毒素与抗体的受控和位点特异性缀合。
类似地,长期以来一直关注的是将来自植物和其他物种的蛋白质毒素与抗体结合,以及这些工程化蛋白质的治疗用途,称为免疫缀合物(immunoconjugates)。 结构指导的方法可以帮助设计蛋白质毒素分子以获得各种稳定性和免疫原性相关的特性。 类似地,作为免疫系统关键调节剂的细胞因子被认为是有针对性作用的有效载荷并且提供了新的工程机会
四、总结
用于治疗的合理的知识和计算建模驱动的蛋白质设计和优化的实用性随着从采用高通量技术获得的信息的激增而扩大,可以在分子水平上欣赏天然生物活性。高性能计算硬件和软件设计的同时改进为利用这些技术优化和应用新型分子建模和模拟技术提供了机会,以解决传统方法难以解决的问题。用于治疗性蛋白质优化的理论计算建模方法的有效开发和使用需要计算科学,化学,生物学和实验评估之间的协作。计算机模拟指导的蛋白质工程方法有助于优化治疗候选物的药效学和药代动力学方面。这种合理的数据驱动的优化程序对于确定治疗作用的形式和开发最佳候选溶液,解决分子稳定性和特异性结合的方面特别有价值。
参考资料
- 《Developability of Biotherapeutics》
