【1.2】肿瘤基因组学
Cancer Bioinformatics: Framework for computational researchers studying the basics of cancer through comparative analyses of omic data. It discusses how key cancer pathways can be analyzed and discovered to derive new insights into the disease and identifies diagnostic and prognostic markers for cancer
人类对恶性肿瘤发病规律、原因和机制的研究已 持续了一个世纪之久, 从细胞癌变起源学说、细胞异 常分裂、染色体易位、基因突变、表观遗传调控到遗 传、进化、发育、代谢的信号调控网络, 提出了许多 的假说, 同时也得到了数十项具有里程碑意义的重 大科学发现, 比较全面地阐述了肿瘤细胞发生发展 的生物学特性和临床转归. 肿瘤细胞的特征是获得 无限增殖和失控性生长的能力, 逃避机体的免疫监 视、以糖酵解代谢方式进行能量代谢、表现为“返祖” 和去分化并以克隆性优势生长进行侵袭和迁移. 肿 瘤细胞的这些生物学特征已脱离了机体正常细胞固 有的基本特性和演化过程, 推测其必然存在决定性 因素和遗传学基础. 因此, 从分子遗传学的角度研究 肿瘤已经成为主流.
临床病人肿瘤突变位点的复杂和多样性
一、从鉴定癌基因到肿瘤基因组学研究
- 最早从分子角度对肿瘤的认识来自David von Hansemann和Theodor Boveri. 他们发现癌细胞分裂 过程中伴随着异常染色体的出现, 由此推测癌症和 异常细胞克隆的形成是由于遗传物质的异常造成 的
- 同时, DNA的损伤和诱导突变导致癌症发生的 现象也佐证了DNA这一遗传物质的异常对癌症的关键作用. 对癌细胞染色体的细致观察发现在慢性髓 性白血病中的9和22号染色体重排(费城染色体)
- 其后, 有研究将癌细胞的DNA转入表型正常的细胞 NIH3T3中, 发现可以使其转化为癌细胞, 从而鉴定 了人类第一个癌基因HRAS, 并将致癌基因突变的研 究深入到众多癌基因、抑癌基因的研究中
21世纪初 Hanahan和Weinberg[5]撰文对基因突变与细胞癌变的 关系进行了系统的阐述, 提出了肿瘤细胞需要具备 的6个特点:
- 持续地获得生长信号;
- 对抗生长 信号失敏;
- 逃避凋亡机制;
- 持续的血管生成;
- 不受限制的复制能力;
- 组织侵袭和远处转移
10年后他们又补充了4个新的特征:
- 基因组不稳 定性;
- 逃避免疫监视的能力;
- 肿瘤介导的炎症;
- 癌细胞能量代谢失调[
20世纪80年代中期, Dulbecco[7]提出获得完整的 人类基因组序列是系统认识癌症驱动基因的必由之 路.
1990年, 人类基因组计划正式启动. 而人类基因 组图谱的获得(2000年获得草图[8,9], 2003年获得完成 图[10]), 为如今肿瘤基因组研究的开展奠定了最重要 的基础
迄今的肿瘤基因组学的进展可以分为两个方面:
- 一是鉴定新的肿瘤相关的突变位点、突变基因和突变 参与的分子调控网络及细胞信号通路,
- 二是从基因 变异的谱系上分析肿瘤发生发展的原因
二、测序技术和生物信息分析方法与肿瘤基 因组变异图谱绘制
对肿瘤基因组学研究而言, 获取高质量且符合医学伦理要求的样本是一个至关重要的环节.
- 多种癌症在临床上取材比较困难, 特别是复发的癌症组织
- 福尔马林固定、石蜡包埋的组织块或切片是肿瘤 基因组研究的重要资源. 但是, 固定和包埋的过程会 对DNA造成损伤, 造成很多处理过程引入的、非肿瘤 特异性的突变[12]; 同时这些样本量相对较少, 经常 满足不了测序的要求
- 全基因组扩增(whole genome amplification, WGA)可以解决样本量不足的问题, 但 同样会造成测序的偏向性[13]、突变的假阳性率提 高[14]和异常的重排事件的出现等[15].
- 另外, 肿瘤的 临床组织样本一般会混杂正常细胞, 包括间充质细 胞、纤维原细胞(肿瘤基质)和渗入组织的淋巴细胞等. 即使没有正常细胞的混入, 肿瘤细胞间携带的突变 也是不均一的. 通过提高测序深度的复发可以一定 程度地弥补这一问题, 但这同样受到测序技术准确 率的限制
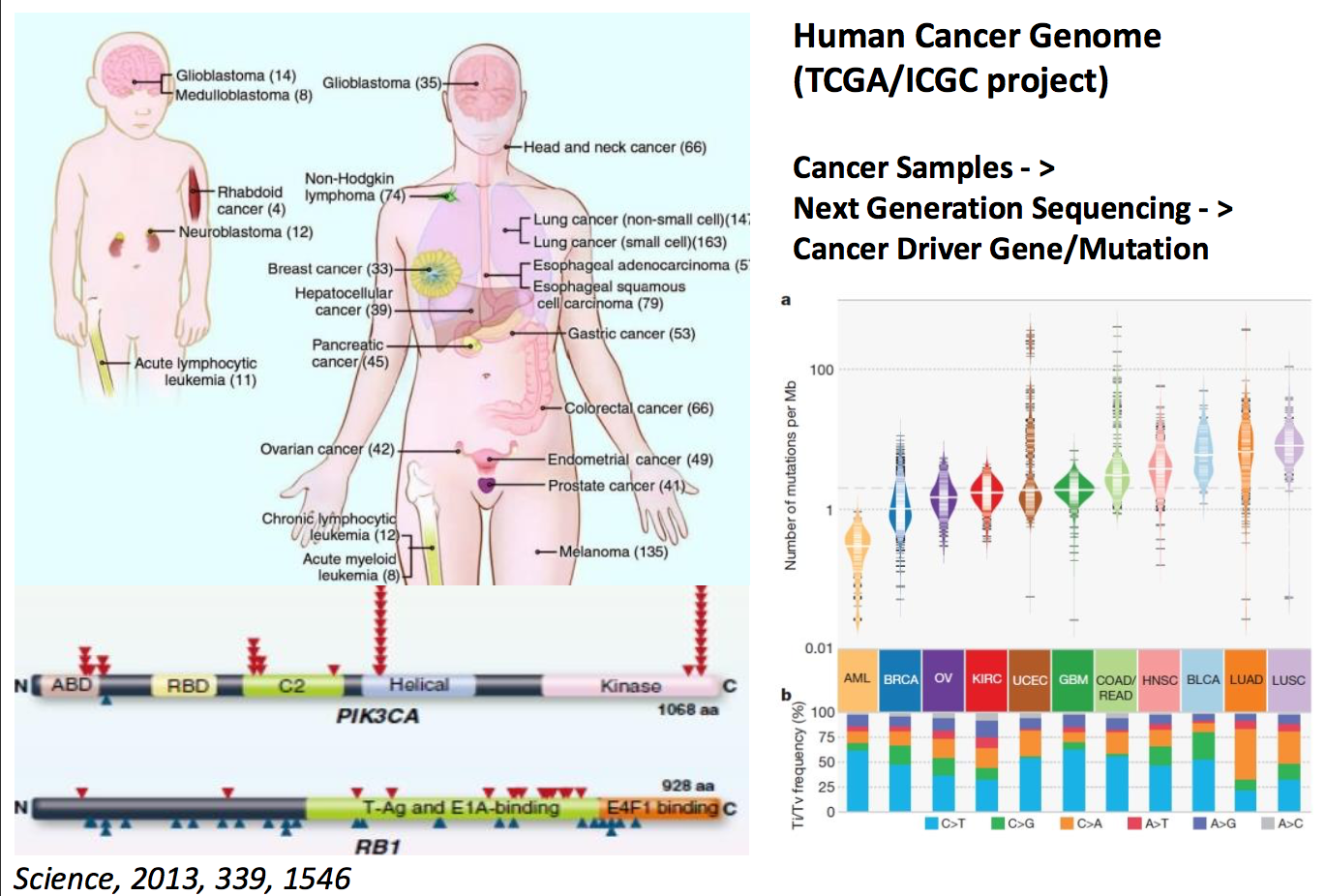
当前肿瘤基因组学研究主要基于第二代测序技 术, 可以使我们对癌症的研究从个别基因的分析和 微阵列研究过渡到全基因组的系统研究. 对肿瘤组 织进行全基因组高深度测序, 并同时对来源于同一 患者的生殖系DNA进行测序作为对照, 这种研究策 略可以使我们一次性获得一个个体完整的体细胞变 异图谱, 包括单碱基突变、结构变异和拷贝数变异 (图1).

但是, 全基因组测序也意味着最大的数据量 和最高昂的实验成本. 序列捕获测序的方法可以对 研究感兴趣的特定DNA区域在相对低成本的基础上 展开更深度的测序, 最常用的是针对所有人类基因 编码区的捕获测序, 即外显子组测序. 核酸探针作为 “鱼饵”将DNA库中的目的序列“钓”出来, 这些探针 可以是DNA[17~20], 也可以是RNA[21]. 另外, 第二代 测序技术也可以从RNA水平对癌组织做出全面的检 测, 即对总RNA、mRNA和其他特定的RNA (如 microRNA, 长的非编码RNA等)反转录形成的cDNA 的测序. 转录组测序对于识别基因间表达的融合现 象非常敏感, 其中一些涉及编码框改变的融合可能 导致癌基因的激活[22]. 也有报道通过转录组测序的 方法识别了与癌症相关的体细胞碱基替换[23]. 最后, 结合DNA和RNA的点突变信息, 我们可以对肿瘤组 织的RNA编辑进行系统的研究
随着肿瘤基因组学研究产生越来越多的数据, 相应的生物信息技术也取得了长足的进步, 重点解 决了以下几个肿瘤研究需要面对的特殊的问题:
- 第 一, 需要同时比较肿瘤组织和正常配对组织的数据来识别稀有的体细胞变异(体细胞点突变发生的比率 一般是人群单碱基多态性发生比例的1/1000);
- 第二, 有能力处理包含大量重排的基因组序列;
- 第三, 有能力处理数据中含量未知的非肿瘤细胞污染和肿瘤细 胞的杂合度.
表1中总结了现有的用于肿瘤基因组分 析的常用软件

三、泛肿瘤基因组学分析
在这里需要重点阐述跨癌种的基因组学研究, 即泛肿瘤基因组学分析. 当今各高发癌症的基因组 变异数据已有很大程度的积累, 比较它们之间的异 同, 分析癌细胞生物学行为更本质的规律, 甚至在分 子水平对癌症做出新的分类系统已经成为可能. 因 此, 泛肿瘤基因组研究已经成为当前在肿瘤基因组 领域的重中之重, 各国生物信息学家无不倾注了巨 大的关注
TCGA与ICGC都有相关的泛肿瘤学的研究
四、基本概念
- Cancer driver mutations:
- Passenger mutations
为什么会有Cancer driver mutation这种说法?
Cancer progression is an example of a rapid adaptive process where evolving new traits is essential for survival and requires a high mutation rate.
Cancer driver mutation 和Passenger mutation的区别
Precancerous cells acquire a few key mutations that drive rapid population growth and carcinogenesis (Driver mutation).
Cancer genomics demonstrates that these few driver mutations occur alongside thousands of random passenger mutations.
五、各种常见的cancer type的缩写
在大量对多种cancer type进行分析的paper中,都会将常见的Cancer type进行缩写 (尤其是在图表中)。
| TCGA(缩写) | Detail(Cancer Type) |
|---|---|
| ACC | Adrenocortical carcinoma |
| BLCA | Bladder Urothelial Carcinoma |
| BRCA | Breast invasive carcinoma |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma |
| CHOL | Cholangio carcinoma |
| COAD | Colon adenocarcinoma |
| DLBC | Lymphoid Neoplasm Diffuse Large B-cell Lymphoma |
| ESCA | Esophageal carcinoma |
| GBM | Glioblastoma multiforme |
| HNSC | Head and Neck squamous cell carcinoma |
| KICH | Kidney Chromophobe |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LAML | Acute Myeloid Leukemia |
| LGG | Brain Lower Grade Glioma |
| LIHC | Liver hepatocellular carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| MESO | Mesothelioma |
| OV | Ovarian serous cystadenocarcinoma |
| PAAD | Pancreatic adenocarcinoma |
| PCPG | Pheochromocytoma and Paraganglioma |
| PRAD | Prostate adenocarcinoma |
| READ | Rectum adenocarcinoma |
| SARC | Sarcoma |
| SKCM | Skin Cutaneous Melanoma |
| STAD | Stomach adenocarcinoma |
| TGCT | Testicular Germ Cell Tumors |
| THCA | Thyroid carcinoma |
| THYM | Thymoma |
| UCEC | Uterine Corpus Endometrial Carcinoma |
| UCS | Uterine Carcinosarcoma |
| UVM | Uveal Melanoma |
四、总结
肿瘤基因组研究产生的大数据和海量的信息如 何利用并使其成为疾病防治的知识将是我们面临的 问题和挑战. 大数据具有4个基本特征: 数据量大 (Volume), 类型繁多(Variety)、价值密度低(Value)、 时效高(V olocity). 因此, 逾越“数据鸿沟”的挑战需 要4种能力即: 预测和判断能力、提炼与排除、集成 验证能力、分类与评价能力. 大数据的核心是预测、 分析更多的数据, 从而可以处理与特别现象相关的所有数据, 例如肿瘤这样的复杂性疾病的健康管理, 而不再依赖随机采样; 数据如此之多, 可能不再热衷 于追求精确度而以明确相关关系和混杂性为主要目 的, 组学和云计算将是主要手段. 同时, 数据共享十分重要, 而且数据越用将变得越有价值. 因此, 组学 和大数据时代, 我们必须改变思路, 发展新的研究模 式和体系, 采用新的方法和技术, 才有可能有新的科 学发现与技术创新.
参考资料
- 《肿瘤基因组学与全球肿瘤基因组计划》
- 沈倩诚 上海宇道生物技术有限公司 《实用肿瘤生物信息学讲座》
