【4.1.2.2】QSAR模型
QSAR,即通过数学方法建立化合物的分子描述符与其生物活性/毒性之间的线性或非线性关系模型,于分子水平阐明结构与生物学及物理化学特性之间的关系。
早在1868年,即有研究者提出化合物的生物活性与其分子结构有某种函数关系;1869年提出几种醇类化合物的硫原子数目与麻醉效果相关;1899年提出化合物的脂溶性对其生物活性有决定性影响;1939年提出一个与SAR相关的方程式;1963年提出将碎片法应用到SAR计算中;1964年提出线性自由能相关模型、相互作用模型;1975年提出分子连接性方法…在以上传统的2D-QSAR研究基础上,1980年出现了CADD技术,继而比较分子场分析法、比较分子相似性指数分析法、比较分子表面分析法等3D-QSAR被提出;再之后,4D-QSAR、5D-QSAR概念相继诞生…但至今,应用最多的还是2D以及3D-QSAR。
2D与3D比较而言,2D计算的时间明显缩短,可作为初筛使用,以减少用于药物开发后期进一步筛选的化合物的数量。而对于QSAR模型的开发,1)需要基于一组类似物的基本化学结构层面来考虑,包含异常值;2)定量关联化学结构变化与生物活性变化之间的关系,以确定最可能决定候选药物生物活性的化学性质;3)基于QSAR结果,来优化现有物质的化学结构,进而继续验证QSAR模型的准确度;4)预测虚拟化合物的生物活性……为了实现以上内容,描述符和方法的选择则至关重要。
一、建模和分子描述符计算
在目前的工作中,按照经合组织和不同研究人员推荐的标准程序进行了 QSAR 分析[16]、[17]、[18]、[19]、[20]、[21]、[22]、[23 ]、[24]、[25]、[26] 。
- 使用 ChemSketch 12 免费软件绘制化学结构,然后使用 TINKER [4]、[12]、[15]中的 MMFF94 力场进行能量最小化.
- 优化的结构被用作计算大量 1-3D、电拓扑、指纹和其他描述符的输入。使用了两个描述符计算软件:PaDEL 2.21 和 e-Dragon。
- 因为,所有计算的描述符 (>18,000) 都不包含重要信息;采用客观特征选择来减少描述符池。在使用 QSARINS-Chem 2.2.1 [16]、[17]、[20]进行主观特征选择 (SFS) 之前,消除了几乎恒定 (>95%)、恒定和高度相关 (|R| > 95%) 的描述符. 这导致仅包含 345 个描述符的集群减少。
- 下一步涉及消除高度深奥的描述符,即无法准确解释或难以根据结构特征解释的描述符[26]。这导致了一组只有 253 个易于解释的描述符。简化集仍然包含广泛的理论分子描述符,这些描述符考虑了不同的结构特征,即,结构 (0D-)、一维 (1D-)、二维 (2D-) 和三维 (3D-),捕捉和放大化学结构的各个方面。
描述符,是以数字形式来体现分子的化学特征,下图概括了描述符的基本定义,其选择主要考虑:1)尽可能使用少数描述符以增加对模型结果的解释;2)降低嘈杂的冗余分子描述符,以降低过度拟合的风险;3)尽可能提供更快且具有成本效益的模型…总而言之,方向就是通过减少输入空间的维度,但不能丢失任何重要信息。而分子描述符,已是QSAR/QSPR建模中最重要的特征之一,且描述符编码的信息通常取决于分子表示的类型和定义的计算算法。包括:拓扑、几何描述符,等等。
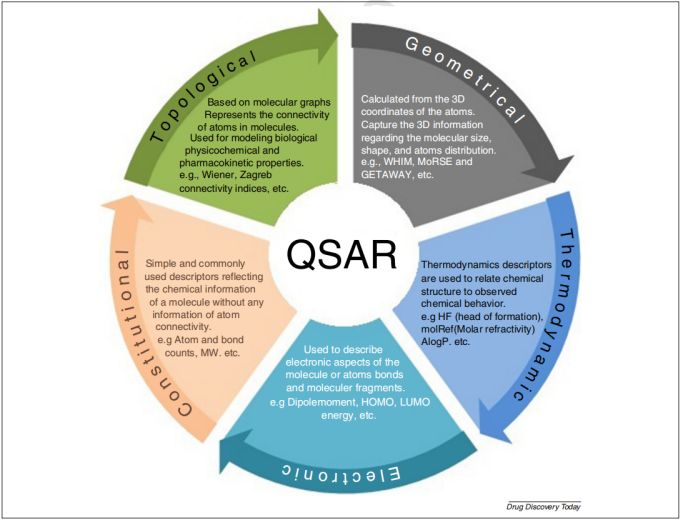
➣ 拓扑指数(TI)
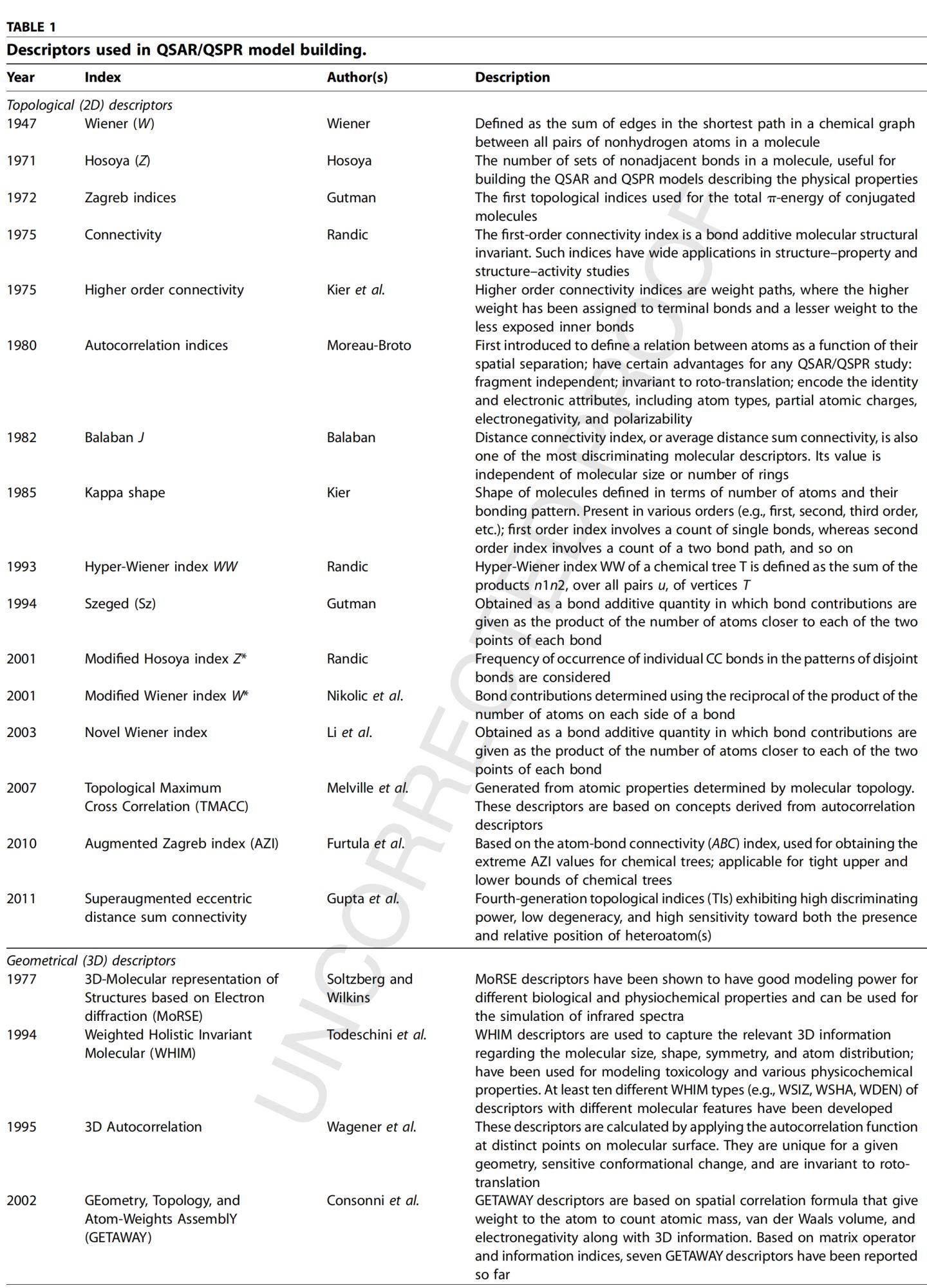
是分子结构的数学描述符,用以反映分子的大小、形状、分支、杂原子、不饱和键等结构特征,从而实现分子结构信息的数值化;在不同理化特性、生物活性和药代动力学特性的建模中具有重要作用;最常用的是Wiener指数、 Connectivity指数、 Kier shape指数、Balaban J指数、以及 Zagreb指数,等。如Wiener指数,是最早的表达化学式构型的拓扑学指数,并已成为QSAR/QSPR研究中最常用的描述符之一。
➣ 几何描述符
根据给定分子中原子的3D坐标计算得出,其对相似的化学结构和分子构象具有丰富的信息和辨别能力;与拓扑描述符相比,可以获得更多信息;但过程需要优化,计算量大。相对而言,对于可以具有多种分子构象的柔性分子,可以获得更多信息;同时,复杂性增加。Ps:下图给出了常用的拓扑和几何描述符形式。
➣ 理化描述符
用来描述物质物理化学特征的参数,如化合物的亲脂性、溶解性和渗透性等。药物的这些特性,在一定程度上可以提高其药效,从而提高产品的临床及市场价值;因此,研究药物的这些特性不仅可以支撑安全性,而且还会大力助力候选化合物的药物发现过程。
- 亲脂性:体内药物转运的关键特性,包括肠道吸收、膜通透性、蛋白质结合和组织分布;重点参数logP(重点关注clogP<5,尤其1~3)。
- 渗透性:依赖于亲脂性,受分子大小、氢键、亲水性和电离度等影响,相关于BCS分类。
- 水溶性:药物开发过程失败的最主要原因之一,与分子大小、刚性、亲脂性相关。
二、QSAR模型
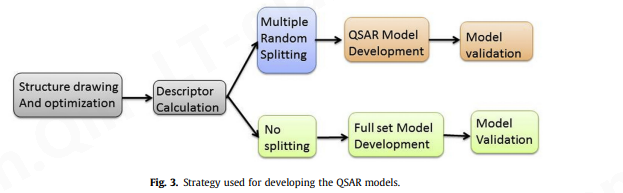
QSAR 分析的首要原则和应用是获得与活性相关的结构特征的最大信息,并在分子实际合成和生物筛选之前预测其所需的活性。因此,为了实现这些目标,在模型生成过程中考虑了易于解释的描述符,并使用划分和未划分的数据集开发了多个 QSAR 模型[19]、[27]、[28]。
- 在选择描述符之前,数据集以随机方式分为训练集(80%)和预测(或测试)集(20%)。
- 采用多重分裂来开发多个 QSAR 模型[12],[15],这样一个分裂的训练集中的分子可能在另一个分裂的训练集中,也可能不在另一个分裂的训练集中。因此,多 QSAR 建模方法确保为控制分子生物学特征的分子描述符获得最大数量和信息。
- QSARINS-Chem 2.2.1的GA(遗传算法)模块用于选择最佳数量和描述符集。为了简单起见并避免过度拟合问题,描述符的启发式搜索仅限于使用 QSARINS-Chem 2.2.1 中的默认设置的四个描述符。
- Q2 loo被用作适应度函数来避免朴素Q 2的问题。QSAR 模型开发中使用的策略总结在图 3 [12]、[15]、[24]。
三、 模型验证
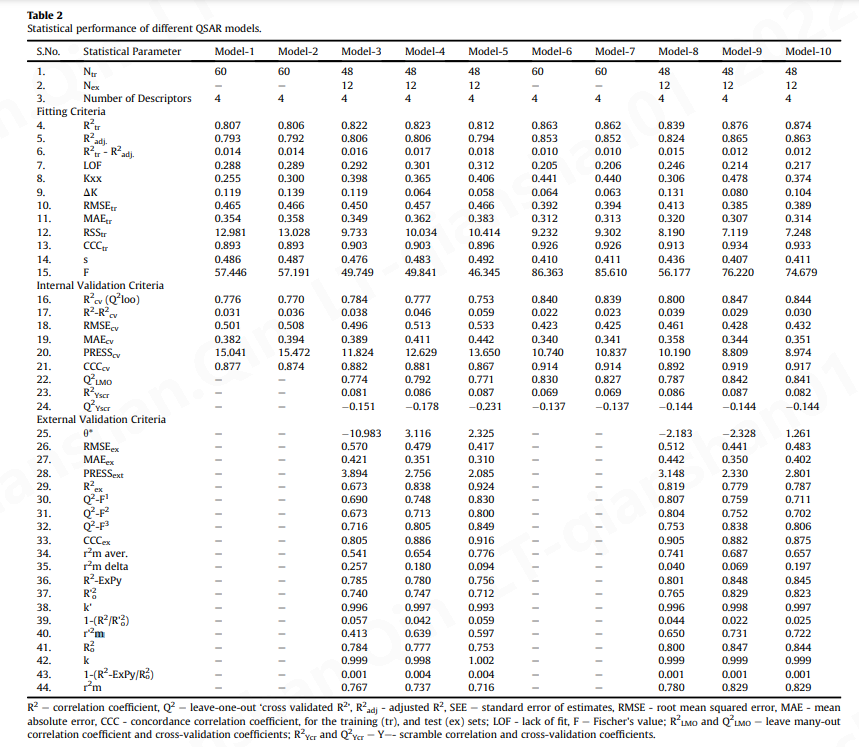
所有 QSAR 模型都需要经过适当验证,以确定其预测能力和实用性。QSAR 模型的统计质量和有效性通过以下方式确定:
- 通过留一法 (LOO,leave-one-out) 和留许多法 (LMO,leave-many-out) 程序进行的内部验证或交叉验证 (CV);
- 使用预测集;
- 数据随机化,即 Y 置乱
- 检查是否满足以下条件[12],[15]:R 2 tr ≥ 0.6,Q 2 loo ≥ 0.5,Q 2 LMO ≥ 0.6,R 2 > Q 2,R 2 ex≥0.6 ,RMSE tr < RMSE cv , ΔK ≥ 0.05, CCC ≥ 0.80, Q 2 - F n ≥ 0.60, r 2 m ≥ 0.6, (1− r 2 / r o 2 ) < 0.1, 0.9 ≤ k ≤ 1.1 或 (1− r 2 / r ' o 2 ) < 0.1, 0.9 ≤ k ' ≤ 1.1, | r o 2 - r ' o 2 | < 0.3,RMSE和MAE接近于零。这些参数的阈值证实了 GA-MLR 模型的稳健性和良好的外部预测能力。
因此,所有具有低内部和外部预测能力的模型随后都被拒绝了。
CV: crosss-validation
为了去除不相关的描述符,需要一个选择标准来衡量每个选择的描述符与分类器输出的相关性,下图描述了描述符的选择流程。该流程已成为开发QSAR模型的基础要求,通过该流程获得的模型,解释性和通用性高度依赖于描述符和目标属性之间的统计关系,这一过程中最好还需要行业专家对数据进行进一步的评价。
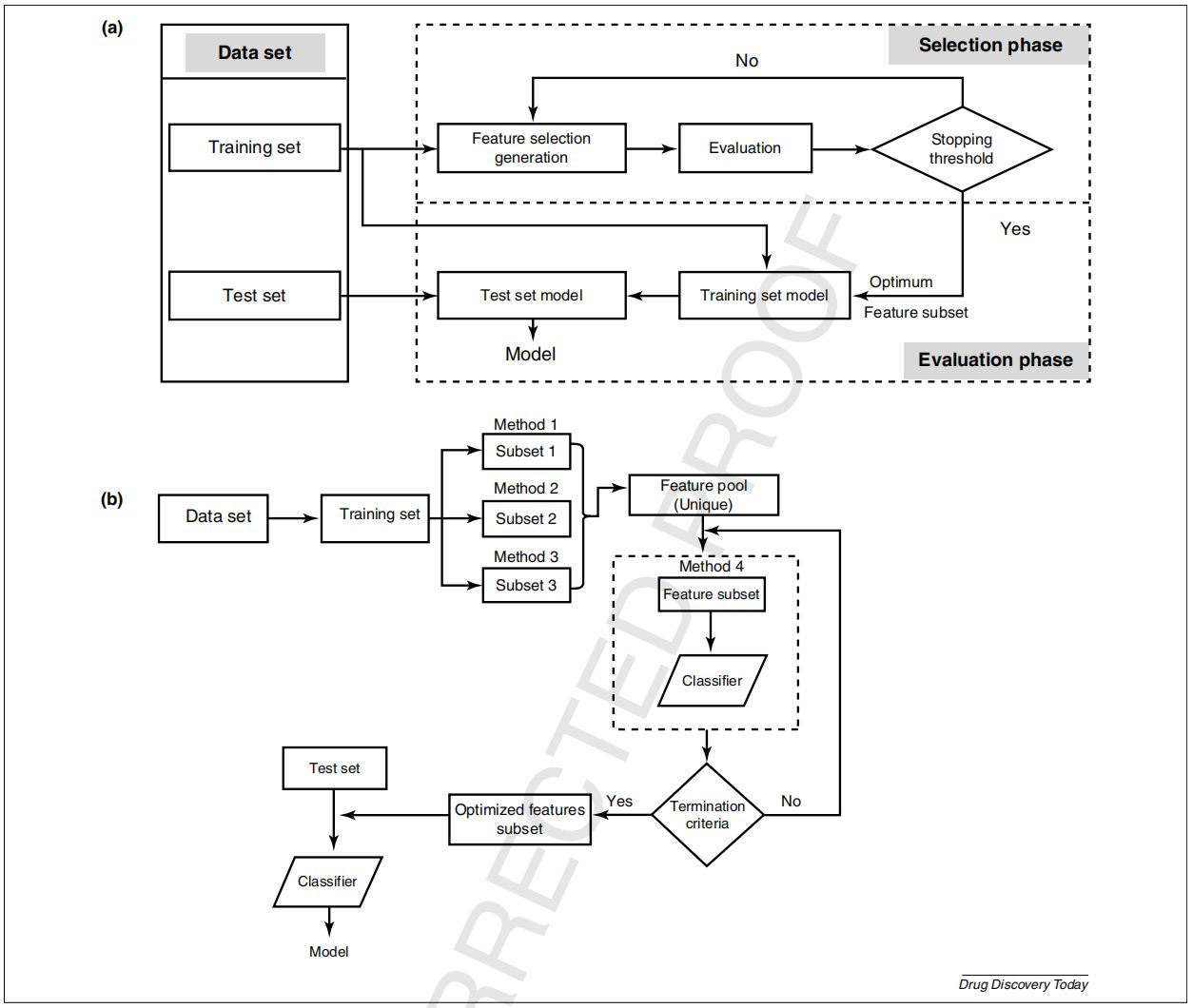
图3.1 特征描述符的选择流程(图片源:见参考文献1)
一项研究中,通过使用主成分分析(PCA)和3D可视化,应用于去乙酰化酶抑制剂的分子描述符,并揭示子空间具有不同的生物活性密度。结果提供的证据表明,某些结构特征对于去乙酰化酶抑制剂的生物靶标活性具有重要意义。下图给出了相关描述符选择的3种策略,即Filter、Wrapper、Embedded。
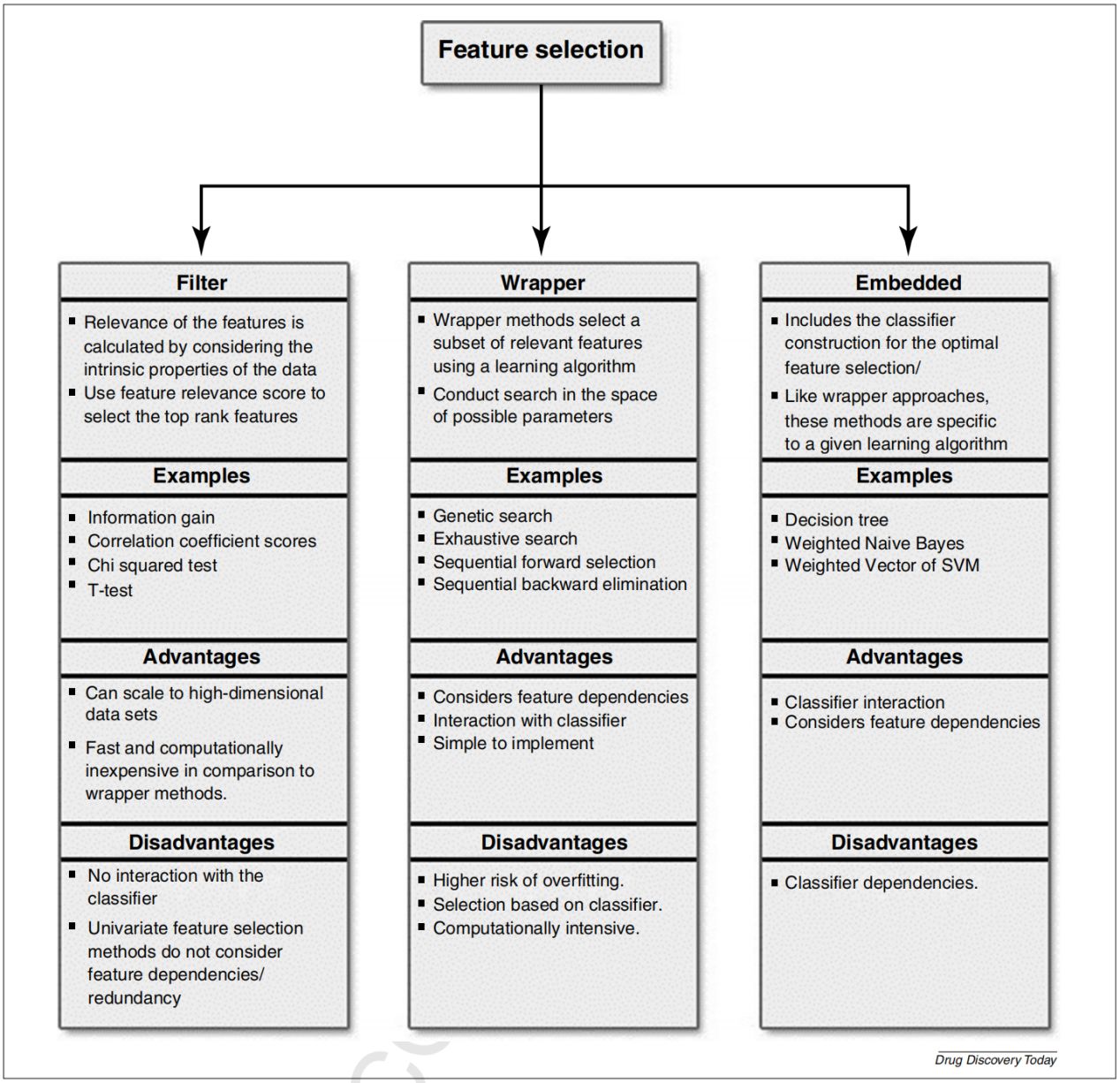
Filter选择描述符的子集作为预处理步骤,独立于归纳算法;优点是简单、快速;缺点是不与分类器相关。Wrapper方法是根据给定子集的分类器函数的误差选择最佳特征子集;与Filter方法相比,性能更好;只使用一个分类器。Embedded对底层分类器的结构很敏感,一种嵌入方法选择的特征可能不适合其他方法。
尽管有许多可用的特征选择技术,但在处理尚未正确理解的复杂性数据的不同方面时,通常建议结合几种方法或者混合方法。如在一项用于开发HIV-1蛋白酶抑制剂的预测模型时,预处理步骤,描述符从1559减少到605,又减少到56,且进一步通过4种不同的机器学习技术,即SVM、k-nearest neighbor(k-NN)、ANN、logistic regression,完成了模型的最终构建。
四、为什么要多个QSAR模型:
然而,这种“平等中的第一”方法具有以下缺点:
- 由深奥的描述符组成的 QSAR 模型,根据结构特征进行适当和现实的描述是非常成问题和具有挑战性的
- 单个 QSAR 模型可能不基于 (i) 训练和测试集的适当组合, (ii) 足够的化学和生物空间,即适当的适用范围,
- 单个 QSAR 模型可能对特定预测集具有高预测性,但对另一个预测集的预测性较差。
为了克服“平等中优先”方法的这些缺点,构建和报告多个模型或共识建模是两种简单、实用且有效的解决方案。
五、另一篇值得被记录的综述
组合文库的虚拟过滤和筛选作为高通量筛选和组合化学的补充方法,最近受到了人们的关注。这些化学信息学技术很大程度上依赖于定量结构活性关系(QSAR)分析,这是一个已建立方法和成功历史的领域。在这篇综述中,我们讨论了建立QSAR模型的计算方法。我们首先概述了它们在高通量筛选中的有效性,并确定了QSAR模型的通用方案。接下来,我们将重点关注构建QSAR模型的三个主要组成部分的方法,即描述化合物分子结构的方法、信息描述符的选择和活性预测的方法。我们介绍了两种成熟的方法以及最近引入到QSAR领域的技术。
5.1 QSAR模型的通用方案
首先,化学结构通常不以明确的形式包含与活动相关的信息。这些信息必须从该结构中提取出来。各种合理设计的分子描述符强调了分子结构中隐含的不同化学性质。
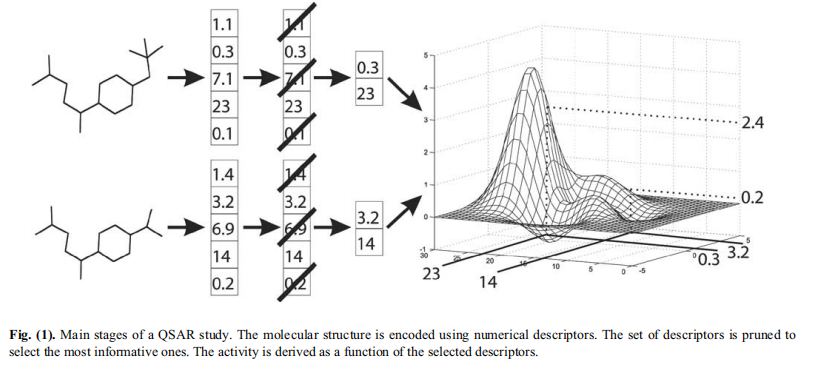
许多应用程序能够生成数百或数千个不同的分子描述符。通常情况下,只有其中一部分与活性显著相关。此外,许多描述符都是相互关联的。这对QSAR分析的有负面影响。大量的描述符也会影响最终模型的可解释性。为了解决这些问题,在QSAR分析中使用了各种方法来将描述符集自动缩小到信息最丰富的方法。
5.2 分子描述符
分子描述符将化合物的结构映射成一组数值或二进制值,它们代表了被认为对解释活性很重要的各种分子性质。根据对分子的三维取向和构象信息的依赖性,可以区分出两大类的描述符。
2.1 二维QSAR
描述符2D-QSAR方法中使用的广泛描述符家族具有一个共同的特性,即独立于化合物的三维取向。这些描述符范围从构成分子的实体的简单测量,通过其拓扑和几何性质到计算静电和量子化学描述符或先进的片段计数方法。
2.2 三维QSAR
描述符三维QSAR方法比二维QSAR方法在计算上要复杂得多。一般来说,它需要几个步骤来获得复合结构的数值描述符。首先,化合物的构象必须从实验数据或分子力学中确定,然后通过最小化能量来进行细化。接下来,数据集中的构象必须在空间中均匀对齐。最后,对各种描述符的浸入构形空间进行了计算探索。一些独立于化合物排列的方法也已经发展出来。
5.3 自动选择相关的分子描述符
对于要选择用于构建QSAR模型的最佳描述符或特征的自动方法,可分为两类[64]。在包装器方法中,描述符子集的质量是通过构建和评估一系列的QSAR模型来获得的。在过滤过程中,不建立任何模型,并使用其他一些标准来评估特征。
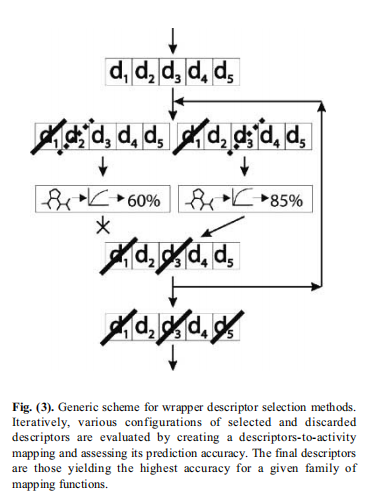
5.4 映射分子结构到分子活性
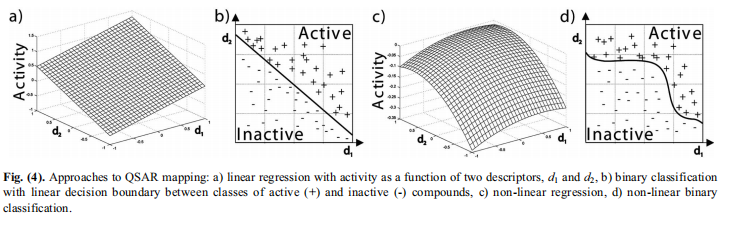
给定所选的描述符,构建QSAR模型的最后一步是推导出活性和特征值之间的映射。简单而又有用的方法将活动建模为描述符的线性函数。其他的非线性方法将这种方法扩展到更复杂的关系。映射方法的另一个重要划分是基于活动变量的性质。在预测一个连续值时,会遇到一个回归问题。当只需要预测某些类别的活性类别时,例如将化合物划分为活性和非活性时,就会出现分类问题。在回归中,因变量被建模为描述符的函数,如上所述。在分类框架中,生成的模型是由一个决策来定义的边界,在描述符空间中分隔类。QSAR映射的方法如图4所示。
5.5 结论
QSAR模型的创建仍然是计算机辅助药物发现的主要任务。一般来说,采用新颖的、更准确的QSAR建模技术并不是一件容易的事情,模型越复杂和优化,在应用过程中就需要越谨慎。结合被检查的数据集的复杂性的增加,这使得QSAR分析成为一项困难的工作。
参考资料
- Journal of Molecular Structure Volume 1130, 15 February 2017, Pages 711-718。QSAR modeling for anti-human African trypanosomiasis activity of substituted 2-Phenylimidazopyridines。 https://doi.org/10.1016/j.molstruc.2016.11.012
- https://zhuanlan.zhihu.com/p/398324971
- Descriptors and their selection methods in QSAR analysis: paradigm for drug design. Drug Discovery Today.
- https://mp.weixin.qq.com/s/BN0XpRmSDJ109H9PXRZa8A
- Dudek, Arkadiusz Z. et al. “Computational Methods In Developing Quantitative Structure-Activity Relationships (Qsar): A Review”, Combinatorial chemistry & high throughput screening 9.3 (2006): 213-228.
