【6.5.1】CRISPR-Cas9介导的基因失活的高活性sgrNAs的合理设计
最近,原核簇状,规则间隔,短回文重复序列(CRISPR,clustered regularly interspaced short palindromic repeats )基因座的组件已重新用于哺乳动物细胞。可以使用单个向导RNA(sgRNA)对CRISPR相关的(Cas)9进行编辑,以产生特定于位点的DNA断裂,但是很少有已知规则可以控制该系统的靶向效力[7,8]。我们创建了一个sgRNA池,将一组六个内源性小鼠和三个内源性人类基因的所有可能靶位点拼接在一起,并通过抗体染色和流式细胞术定量评估了它们产生靶基因无效等位基因的能力。我们发现了改善活性的序列特征,包括化脓性链球菌Cas9的原间隔物相邻基序(PAM)的进一步优化。 1,841个sgRNA的结果用于构建 sgRNA活性的预测模型,可改善用于基因编辑和基因筛选的sgRNA设计。我们提供了一种在线工具,可针对任何感兴趣的基因设计高活性sgRNA。
一、前言
当引入哺乳动物细胞后,Cas9:sgRNA复合物产生序列特异性双链(ds)DNA断裂,该断裂通过易错的非同源末端连接(NHEJ,error-prone nonhomologous end-joining )途径修复,通常通过移码等位基因的产生导致基因失活。 最近,我们和其他人已经证明,CRISPR技术可以用于哺乳动物细胞中大规模的基因筛选[7,9-11]。 这些筛选的命中基因在靶向同一基因的不同sgRNA之间显示出高度的一致性,高于在RNA干扰(RNAi)筛选中通常看到的一致性。 这表明敲除等位基因的表型后果可能比RNAi诱导的敲除程度不同更一致,并且尽管可以检测到sgRNA的脱靶效应,但很少见,足以使多个靶向同一sgRNA的sgRNA高度一致 基因,促进真正阳性命中的提名。
据我们所知,到目前为止,所有基于CRISPR的遗传筛选,都依赖于对细胞毒性药物耐药的阳性选择或基于必需基因消耗的阴性选择。尽管产生了令人鼓舞的结果,但这些测定法都涉及强大的选择性压力,即使只有中等比例的接受特定sgRNA的细胞经历了完整的基因失活,这些测定法也有望显示出强大的信号。许多未来的筛选,特别是那些无法通过提高存活率或增殖来衡量筛选成功的筛选,将需要很大一部分经sgRNA处理的细胞才能完全敲除,因为没有敲除等位基因,杂合敲除或亚型等位基因的细胞会稀释测定信号。迄今为止的研究表明,尽管sgRNA的活性可能很高,但sgRNA产生无效等位基因的能力之间存在显着差异。因此,最大化sgRNA功效的设计标准非常有用,可以改善筛选文库,还可以用于规模较小的基因编辑实验,这通常需要研究人员首先筛选多个sgRNA的活性。
因此,我们试图发现目标位点内和周围的序列特征,从而预测sgRNA的功效。为了发现通常适用的规则,我们针对多个基因靶标测试了多种sgRNA。我们的策略是针对大细胞群体中的细胞表面标记,每个细胞传递一个sgRNA,然后通过荧光激活细胞分选(FACS)分离出完整的(双等位基因)敲除细胞,从而分离出活性最高的sgRNA。我们设计了针对所有外显子和所有侧翼内含子序列的一组小鼠基因的sgRNA,这些基因位于化脓性链球菌Cas9要求的NGG PAM之前的所有20个核苷酸(nt)靶位点上,并添加了大量阴性对照sgRNA(图1a和补充表1)。如前所述,这些sgRNA被作为一个池克隆到慢病毒载体中,该载体同时传递CRISPR相关蛋白(Cas)9,赋予嘌呤霉素抗性并表达sgRNA。将靶向三个人类细胞表面标记的编码序列,并还包括阴性对照的第二个库,分别克隆到仅表达sgRNA的慢病毒载体中(图1a和补充表2)。
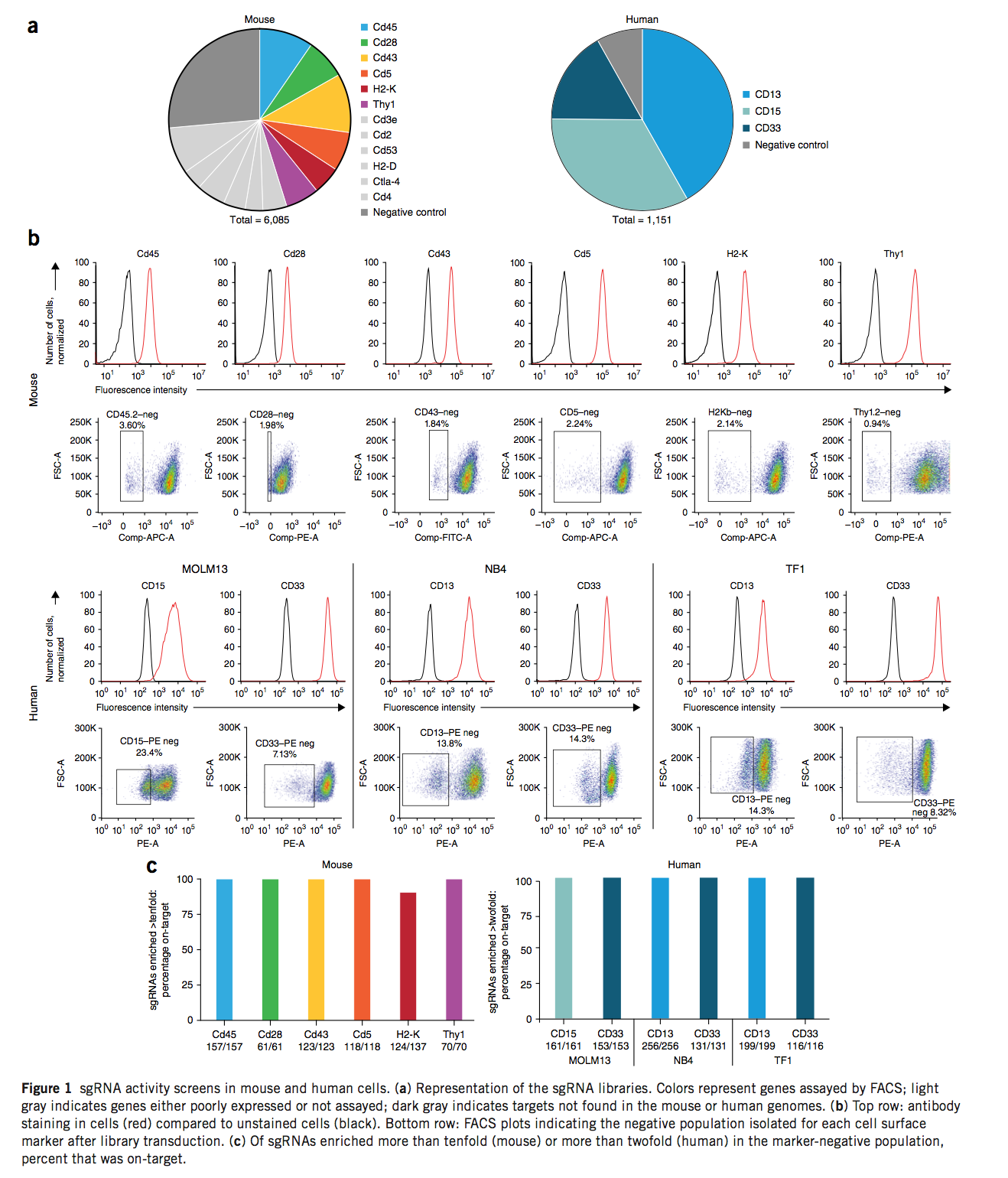
我们用小鼠sgRNA库转导了小鼠胸腺细胞系EL4细胞。转导后第9天,我们对9种细胞表面标志物中的每一种进行了细胞染色,并通过FACS分析了它们。内源性Thy1,H2-K,Cd45,Cd43,Cd28和Cd5表现出良好的标志物阴性细胞群解析度(图1b),而Cd2,Cd3e和Cd53的表达较差,无法进行后续分析(补充图1)。为了评估人类基因靶向,我们准备了 通过传递Cas9并赋予杀稻瘟菌素抗性的载体进行转导,从而获得三种人类急性髓样白血病细胞系MOLM13,NB4和TF1。我们在这些多克隆品系中确认了Cas9的活性(补充图2),然后在人sgRNA库中进行了转导,并在转导后8 d收集了标记阴性人群。在其中一个,两个和三个细胞系中分别评估CD15,CD13和CD33。对于所有12种分选的细胞群,对基因组DNA进行PCR,然后进行下一代测序,确定了导致目标蛋白质完全丧失的sgRNA(补充表2和3)。
我们首先检查了靶向每个基因的sgRNA的敲除特异性。在小鼠池中,我们观察到每个基因的61-157个sgRNA均富集了至少十倍,然后将每个分类的人群中sgRNA的丰度标准化为未分类人群中的起始丰度。在人类库中,每个基因的116-256个sgRNA经过相似的标准化至少富集了两倍;将较低的阈值用于此库,因为每个基因占整个库的较大部分。在12种分类标记阴性的人群中,有11种中,高活性sgRNA是靶向分类标记的,即“靶向” sgRNA,显示sgRNA的出色特异性以及这种基于FACS的reads(图1c)。对于H2-K,所有十倍富集的脱靶sgRNA都包含在池中以靶向H2-D,H2-D是一种在EL4细胞中未表达但与H2-K序列高度相似的基因。所有这些靶向H2-D的sgRNA都具有至少17个与H2-K互补的nt,其中11个仅相差一个核苷酸(补充表4)。正如先前关于sgRNA特异性的研究所期望的那样,保留了活性的单碱基错配在sgRNA序列的5’一半处更为常见。使用广泛使用的脱靶评分算法,许多这些脱靶sgRNA均被评定为可能的脱靶匹配。但是,有几个接收到的脱靶得分较低,这表明脱靶预测可能有改进的空间。
接下来,我们检查了跨细胞系的靶向CD13或CD33的sgRNA活性的一致性。 我们在四个成对的跨细胞系比较中观察到了高度相关的sgRNA活性,这表明sgRNA活性的相对水平可以在整个细胞环境中普遍存在(图2a和补充图3)。 为了进一步验证以合并筛选形式获得的结果,我们以阵列形式重新测试了靶向三个基因的17个sgRNA的活性,并观察到这两种测定法之间的良好对应性(补充图4a和补充表5)。 然后,我们检查了由17种sgRNA引起的DNA损伤谱。 如预期的那样,我们发现移码等位基因在标记阴性人群中更丰富的sgRNA更常见(补充图4b)。
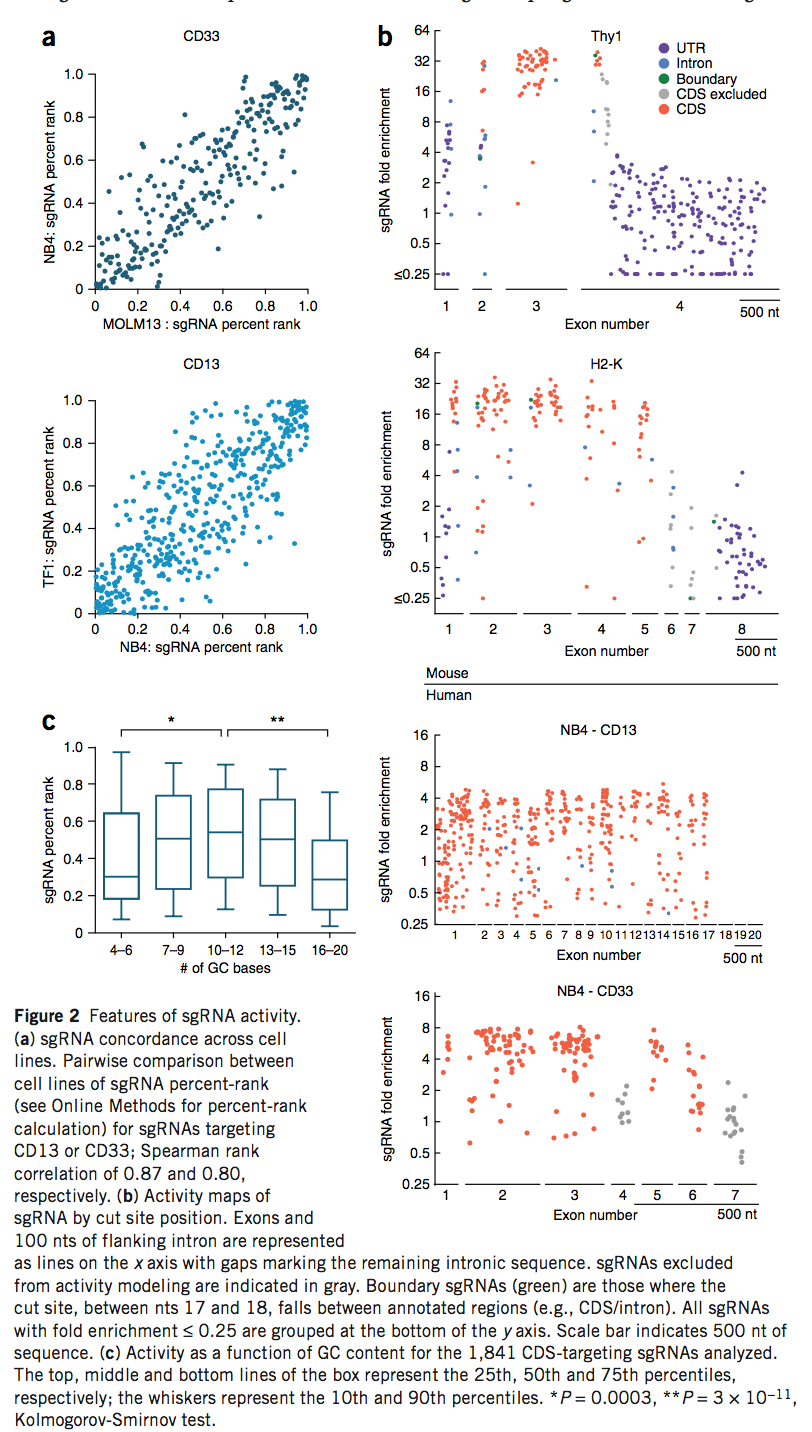
对于所有九个靶基因,我们通过切割位点的位置注释每个sgRNA,以确定基因中靶位点的位置如何与其功效相关(图2b,补充图5和补充表6)。一些外显子不包含活性sgRNA靶标,表明这些外显子在测定的细胞系中不表达(补充图5)。正如预期的那样,我们观察到靶向靠近C’端的sgRNA的活性降低,因为接近蛋白质末端的移码突变不太可能破坏表达(补充图6)。基因特异性模式也出现了。例如,CD15的N’末端是一个不太有效的靶位点,可能反映了局部染色质结构。这些结果表明,尽管通常将宽范围的编码序列(CDS)(wide-range of the coding sequences)用作目标位点,但基因特异性特征可能会引起例外。在文库设计的背景下,针对每个基因一个以上的位点应该有助于弥补基因特异性的局限性。
接下来,我们检查了靶向小鼠库中非编码区的sgRNA的活性。我们看到55%的sgRNA在敲除细胞中富集了十倍以上,而预期的切割位点恰好位于外显子-内含子边界。活性随着与最近的CDS距离的增加而迅速下降:在50个sgRNA中,只有2个具有预期的切割位点6 nts或距离CDS更远,其富集程度超过10倍(补充图7)。最后,我们观察到,靶向5’和3’非翻译区(UTR)的sgRNA效率极低:119个5’UTR靶向的sgRNA中有1个,而1,044个3’UTR靶向的sgRNA中有0个在TAR-中至少富集了十倍。获得基因阴性的细胞群体。这些结果表明,sgRNA通常应设计为靶向CDS,尽管破坏剪接的靶位点可能是有效的,并且在希望重新引入CDS(例如用于表型拯救实验)时可能特别有用。
为了确定与活性相关的sgRNA的序列特征,我们集中于靶向CDS的sgRNA的子集。我们消除了在广泛无效的靶区域(例如,靠近C’末端或明显缺乏外显子表达的区域)中的所有sgRNA,从而产生了1,841个sgRNA,通过每个基因中的等级百分数进行归一化(补充表7)。我们检查了靶链作为活性的函数,与先前观察到的对反义链的轻微偏爱相比,没有发现统计学上的显着差异(补充图8)(参考图7)。此外,我们观察到,具有低或高GC含量的sgRNA的活性往往较低(图2c),如先前报道的7,8。
接下来,我们在整个sgRNA和侧翼靶序列的每个位置检查了活性sgRNA的核苷酸偏好。具体来说,我们在相同基因靶标的20%活性最高的sgRNA中寻找具有给定序列特征的sgRNA的统计富集或耗竭,因为这些高活性sgRNA最为令人关注(图3a和补充表8) 。在sgRNA序列中,最显着的差异出现在位置20(紧邻PAM的核苷酸)上。与以前的观察结果一致,我们发现鸟嘌呤是最优选的,在我们的数据中,胞嘧啶是非常不利的[7,8]。此外,我们在第16位看到了对胞嘧啶和鸟嘌呤的偏好,这是最近对Cas9结合亲和力进行的全基因组分析确定的种子区域的最后一个核苷酸。与Wang等人进一步达成一致,在sgRNA的中间位置始终对腺嘌呤具有一致的偏爱,而在位置3处的胞嘧啶却是不利的。
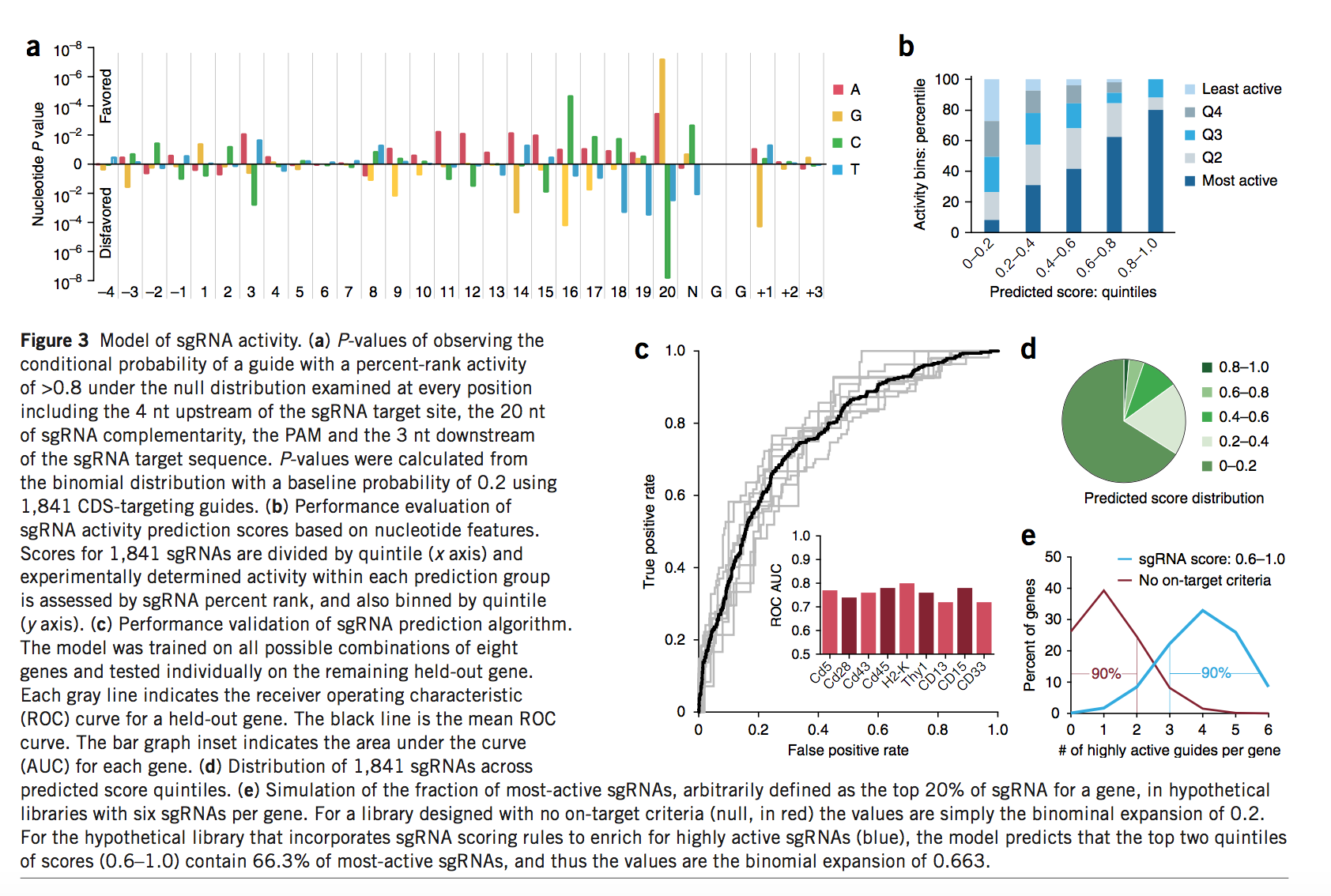
值得注意的是,我们还观察到偏爱PAM的可变核苷酸,其中偏爱胞嘧啶而不利于胸腺嘧啶。最近在斑马鱼中也观察到在这个位置对胞嘧啶的偏爱。我们和其他人观察到的对胸腺嘧啶偏向20-nt sgRNA靶位点3’端的现象先前已从sgRNA表达的角度进行了解释,因为RNA聚合酶III终止于富尿嘧啶的区域,且转录序列紧接在下游20nt的靶向序列是富含尿嘧啶。但是,无法扩展该机制来解释PAM中对胸腺嘧啶的偏倚,因为该胸腺嘧啶是DNA靶位点的特征,并且不包含在sgRNA转录物中。此外,我们观察到对PAM的3’鸟嘌呤有很强的偏倚,表明CGGH的扩展PAM序列最适合用于化脓性链球菌Cas9改造哺乳动物细胞中的dsDNA断裂。实际上,CGGT PAM的靶标中有39%位于活性最高的五分位数,而最低的五分位数只有11%。相反,TGGG的PAM序列最不理想的目标中有42%位于最低活动度的五分位数中,而只有8%位于最高活动度的五分之一中。
我们通过训练逻辑回归分类器来建立sgRNA活性的预测模型,以使用序列特征区分每个基因的sgRNA活性最高的五分位数。我们使用来自所有九个小鼠和人类基因的数据来确定序列特征权重,以进行活性预测(补充表9)。得分最高的五分位数的80%由活性最高的sgRNA组成,包含的活性最低的sgRNA最少(图3b)。相反,得分最低的五分位数包含活性最低的sgRNA和活性最高的sgRNA的最小部分。我们提供了一个使用此模型的简单网络工具,可针对任何感兴趣的序列生成sgRNA分数( http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design )。
为了确保该模型可以跨基因泛化,我们首先通过在保留其余基因的同时对八个基因进行训练对它进行交叉验证,然后该模型准确地预测了所有九个保留基因的活动(图3c)。同样,仅由小鼠库中959个sgRNA决定的碱基偏好与使用完整的1,841-sgRNA数据集获得的偏好紧密融合(补充图9)。值得注意的是,这9个基因的GC含量和长度范围很广,并且没有任何明显的序列同源性,这与在这些基因中sgRNA没有交叉反应的观察一致(图1c)。这些分析表明,数据集足够大,足以使模型收敛于基本偏好的一致模式。
我们使用来自早期筛选的A375细胞(人类黑素瘤系)中的活力影响的数据,进一步验证了该模型针对一组针对414个基因的1,278个sgRNA的通用性。我们检查了先前确定为所有细胞类型(例如蛋白酶体,核糖体)中的必需基因高度富集的功能类别,并分析了在该生存力筛选中具有多个靶向sgRNA且随时间耗尽的基因子集。然后,我们将针对这414个基因的sgRNA的预期功效评分与其在屏幕上观察到的耗竭进行了比较。与我们对FACS蛋白敲除测定的观察结果相似,我们看到预测分数的最高五分位数包含高活性sgRNA的最大比例,而分数最低的五分位数具有最低活性的sgRNA(补充图10和补充图)表10)。对靶向414个基因的1,278个sgRNA的活性的这种预测,以及在所有sgRNA的9个基因的基本偏好中观察到的高一致性,表明此处介绍的模型广泛用于预测高活性sgRNA。
对于筛选方法而言,提供良好的基因组覆盖范围的有效sgRNA文库具有最重要的意义,因此,与更准确地模拟所有sgRNA的活性相比,我们更关心的是正确识别活性最高的sgRNA。结果,此处给出的评分系统对预期活动进行了严格评分:只有5%的sgRNA得分为0.6或更高,而大多数sgRNA(包括许多实验上高度活跃的sgRNA)获得的得分<0.2(图 3d)。因此,该模型最强大的应用是作为sgRNA设计工具,即选择一些得分最高的sgRNA以获得最可能高效的工具。现有的全基因组文库尽管旨在避免脱靶位点,但并未加入任何增强靶上活性的标准[7,9-11]。例如,每个基因设计有六个sgRNA且没有任何靶标活性标准的文库,在90%的基因的最高活性五分之一中将包含两个或更少的sgRNA,而提出了以增强活性的标准设计的文库在90%的基因中,活性最高的五分位数中至少有3个sgRNA(图3e)。
最近已确定局部染色质结构是影响Cas9找到PAM并开始将DNA与sgRNA种子区(seed region )结合的能力的主要因素。我们发现促进活性的序列特征适用于不同的细胞环境,这表明基于Cas9的DNA靶向中涉及的某些步骤受靶位点和sgRNA序列的固有特征支配。我们推测,即使采用最佳设计规则,某些细胞环境或序列特性仍可能使某些基因难以利用当前的CRISPR技术有效靶向。在这些情况下,RNAi技术可能为探测基因功能提供了更好的选择。
在这里,我们定量分析了数千个sgRNA的活性,以揭示可调节Cas9结合DNA,切割靶位点并产生无效等位基因的能力的序列特征。以前类似的方法已应用于RNAi敲低[22,23]。我们使用目标蛋白水平的直接测量方法来对sgRNA活性进行分类,而不是对通常无法区分双等位基因失活与单倍功能不全的表型结果进行分类。确实,将大量的sgRNA与蛋白质敲除功效的定量分析相结合可能允许检测PAM区域中以前没有观察到的偏好。我们发现了可预测sgRNA活性的序列特征,基于这些特征开发了定量模型以优化sgRNA活性预测,并创建了使用该模型进行sgRNA设计的工具。序列特征模型可概括为人类和小鼠起源的9个不同基因和4个细胞系中的每一个,以及全基因组增殖筛选中414个命中基因的sgRNA活性。通过合并其他数据集,活动读数和建模方法,将有可能进一步完善这些活性预测,确定哪些机械步骤驱动序列偏好并确定影响活动的其他因素。此处显示的与Cas9:sgRNA复合物的靶向活性相关的序列特征,将使CRISPR技术能够更有效地应用于编辑基因组和探针基因功能。
二、方法
实验方法略
2.1 数据处理和分析
通过计算每种实验条件下每个sgRNA的独特reads数量来处理Illumina测序读段(补充表2和3)。对于每种sgRNA的每个样品,通过将单个sgRNA的读数数除以该样品中sgRNA的读数总数,乘以一百万,相加一,然后进行log2转化,确定“Reads per Million”。然后将相同实验条件下的多个样品取平均值。
为了分析合并的筛选数据,通过从标记阴性人群的丰度中减去未分选人群的丰度,计算每个sgRNA的对数倍数变化值。我们排除了运行4个或更多胸苷的所有sgRNA,因为这将导致过早的转录终止。我们还排除了所有未分类人群中小于百万分之32个读段的sgRNA。我们目视检查了作为切割位点位置的函数的活动图,以从我们的预测模型中排除针对通常活性较低的区域的任何sgRNA,即使这意味着要排除某些具有较高活性的异常sgRNA,以确保我们不会由于其目标位点,而不是其固有的潜在功效而被错误分配为低活性的序列,污染了我们的建模数据集。
偏离目标得分(补充表4)是从 crispr.mit.edu, accessed April 24, 2014。数据来自Hsu等,用于计算分数,如网络服务器上所述
2.2 sgRNA活性预测模型
在每个基因中,首先对通过的sgRNA进行排名,最好的sgRNA的排名为1。然后将这个数字除以sgRNA的总数,然后从总数中减去sgRNA的百分比。这将导致基因的最差sgRNA的等级秩为0,而最佳sgRNA的等级秩接近1。对于在多个细胞系中测定的基因,将等级秩的平均值取平均值。
用于预测的特征是通过30 mer靶位点的位置索引的单个核苷酸和所有相邻核苷酸对。我们还包括了20 nt sgRNA中的Gs和Cs计数。由于观察到的GC含量和功效之间存在非线性关系,因此还合并了两个GC计数功能:一个用于小于10的偏差,另一个用于大于10的偏差。为了给每个核苷酸特征提供独立的权重,用一热编码来表示核苷酸特征空间。
因为所有特征(包括120个单核苷酸特征,464个二核苷酸特征和两个GC计数特征)都已确定,所以我们合并了特征选择以选择具有最佳泛化误差的特征子集。使用python模块scikit-learn中实现的L1正则化线性支持向量机(SVM),来生成根据L1范数惩罚的功能集。给定SVM的功能集,对逻辑回归分类器进行了训练,以区分其余每个基因的sgRNA的前五分位数。我们通过训练八个基因的数据并预测剩余基因的数据来交叉验证模型。特征选择步骤在训练数据的嵌套分层交叉验证循环中运行,其中,对于八种训练基因中的每一种,每个折叠都排除了相等比例的sgRNA。选择L1范数罚分以使嵌套循环中的平均保持AUC最大化。我们还使用了留一基因的交叉验证来衡量模型的性能,尽管留一基因的输出是更通用的泛化性能指标。验证后,我们使用所有可用数据(补充表9)训练了最终模型,该模型仅使用586个功能中的72个,包括两个GC计数功能。
补充表9中显示的模型权重可用于轻松计算sgRNA得分。 指南仅包含所有功能的子集,通过一热编码将其表示为二进制变量。 令特定指南sj的特征i的模型权重为wij,即截距int。 然后通过逻辑回归得出sgRNA分数f(sj)为:

模型得分f(sj)将落在[0,1]范围内,并且较高的值表示较高的活性。
2.3 A375生存力数据分析。
为了分析致死性(lethal)sgRNA的A375筛选数据,我们感兴趣的是生成一组假阳性尽可能少的sgRNA,并且对捕获所有真实的阳性基因不感兴趣。 从64,751个sgRNA的文库开始,我们应用了许多过滤器来改善数据质量:(i)在早期时间点删除了少于百万分之八的读数的任何sgRNA。 (ii)去除任何包含四个或更多胸苷的游动的sgRNA; (iii)仅检查了针对已经确定为生存力必不可少的基因的sgRNAs。 (iv)需要至少两个靶向该基因的sgRNA保留在数据集中。
这产生了1,278个sgRNA的列表(补充表10)。 我们从单个sgRNA引起的消耗中减去基因平均消耗,以产生每个sgRNA的基因标准化活性。
参考资料
- Doench, J.G. et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267 (2014).
