【4.0.1】RNA二级结构的全基因组分析
现在越来越明显的是,RNA 结构不仅对特定类别的 RNA,如核糖开关、小 RNA 和酶 RNA(核酶),而且对基本上所有的 RNA,包括 mRNA 和长链非编码 RNA (lncRNA),都起着重要的调节作用。碱基配对变化在基因调控中发挥简单但强大的作用,部分是通过隔离或暴露可与其他 RNA、蛋白质或其他细胞成分相互作用的核苷酸。因此,RNA 结构可以被视为遗传密码的另一个先前隐藏的层,我们才刚刚开始理解。
在过去五年中开发的将 RNA 结构探测与高通量测序技术相结合的实验方法已经揭示了全基因组范围内的 RNA 二级结构。这些方法的应用还揭示了结构如何为所有类型的 RNA,特别是 mRNA 提供功能的一般原则。尽管第一代此类方法仅限于体外应用 (118),但从 2013 年开始开发的新方法能够在体内进行全基因组结构探测 (22)。
二、影响 RNA 结构的参数
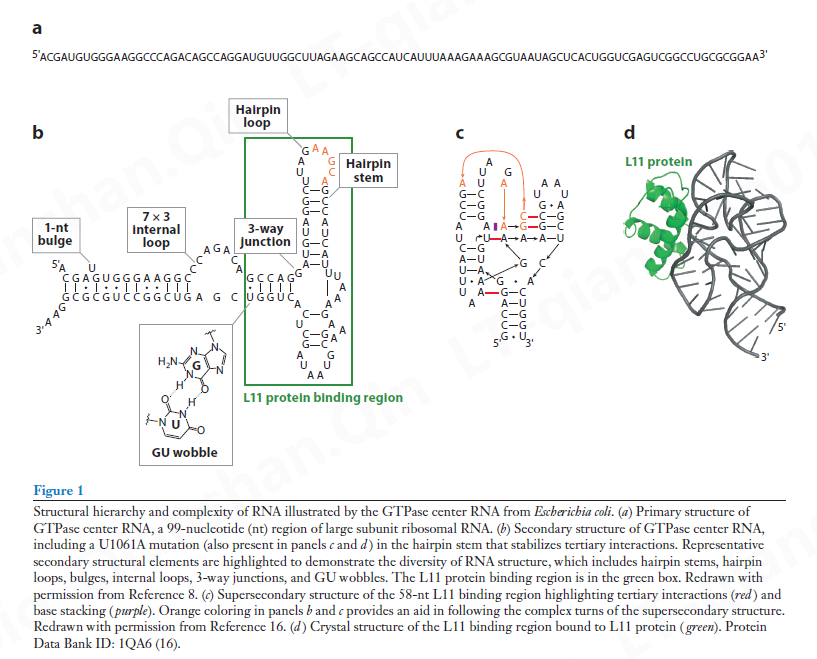
在最基本的水平上,RNA 二级结构由碱基配对和不配对的核苷酸组成,从中产生发夹、凸起和假结的茎和环结构,其中发生反式环碱基配对(图 1)。碱基对要么是规范的(例如,A-U 和 G-C),要么是非规范的(其中 GU 摆动是最常见的例子)。每个碱基对都有一个相关的上下文自由能。根据这些基础知识,以及对短寡核苷酸结构熔解的广泛实验测量,开发了一组最近邻参数,即所谓的Turner rules (76, 128)。这些规则可以应用于给定的 RNA 序列,以通过自由能最小化来预测其二级结构,它们构成了 RNA 结构预测的计算机计算方法的基础,如几个广泛使用的程序中所提供的,例如 RNAstructure (87)、 ViennaRNA 包 (66) 和 Mfold (73)。升高的温度倾向于展开 RNA;因此,RNAstructure 程序 (69) 和 ViennaRNA (66) 已将温度作为结构预测的一个组成部分。
然而,计算机预测无法预测复杂 RNA 聚合物的实际结构 (18)。 尽管最流行的计算机方法采用自由能最小化,但这种方法并未完全由实验数据参数化。 事实上,考虑到影响 RNA 结构的变量的数量,这种参数化基本上是一项不可能完成的任务。 例如,阳离子强烈影响 RNA 结构,尤其是倾向于促进二级和三级折叠的 Mg2+,以及倾向于展开 RNA 的重金属。 此外,pH、相容溶质和拥挤度都对 RNA 结构产生各种复杂的影响。 尽管这些变量可以在体外明确定义,但它们在生命系统中的浓度通常仍然难以定义和/或高度调节,因此在试管中精确复制体内条件仍然是不可能的。
活细胞增加了更多层次的复杂性。与其他分子的相互作用,例如蛋白质和小分子(例如,在上述核糖开关中起作用的那些)与 RNA 的结合,会对 RNA 结构产生深远的影响。酶促反应可以通过 RNA 的共价修饰来影响 RNA 结构,这可以改变 RNA 的稳定性 (38, 90),或者在 RNA 解旋酶的情况下通过解旋来影响 RNA 结构。此外,所有生命系统都消耗能量以非最低自由能状态存在,但自由能最小化方法假设系统中的所有 RNA 都已达到平衡。事实上,有大量证据表明,一些 RNA 折叠成非最小自由能构型的生物学相关结构 (15, 127)。最后,如果试管中没有非常具体的实验条件,例如高浓度的 Mg2+,即使是高度结构化的 RNA 也倾向于存在于多个状态中,而不是填充单个显式结构。即使对于纯粹的计算机结构预测,对这种生物学相关的群体异质性进行建模也是当前的主要挑战。
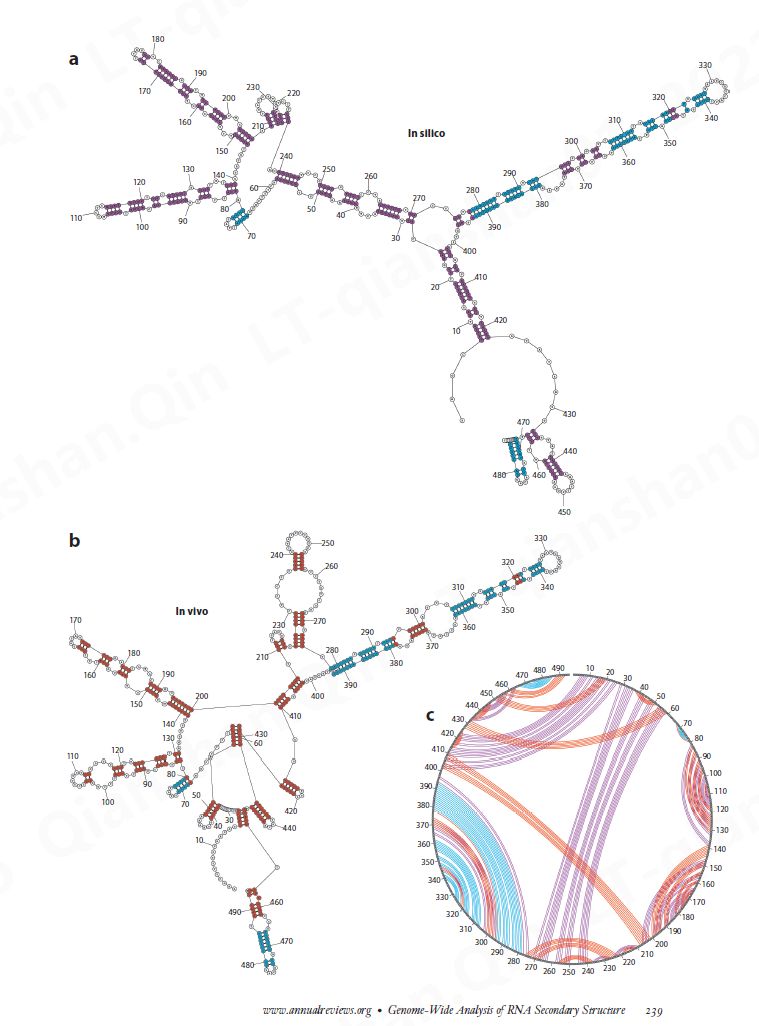
由于上述所有原因,改进对生命系统中 RNA 结构的预测需要开发用于查询 RNA 结构的湿台方法,并且需要推导一般的结构-功能原理,要求此类方法适用于全基因组。此外,由于不可能在体外忠实地概括生命系统的所有物理化学和酶学方面,因此最具生物学信息量的结构探测方法将是那些可以在体内应用的方法。如上所述,仅使用热力学参数(所谓的计算机方法)无法很好地预测大多数 RNA;相反,实验数据被纳入以限制计算机预测算法和产量预测结构 (75, 101)。图 2 说明了当 RNA 结构仅在计算机上预测时与当它受到来自 Structure-seq (21, 22) 的折叠实验信息的限制时可能出现的巨大差异,这是讨论的新的体内结构探测方法之一在本文中。

三、全基因组实验性 RNA 结构探测的方法
技术发展在 RNA 生物学和化学领域的许多里程碑式发现中发挥了关键作用。 例如,RNA 和 DNA 的测序是通过 RNA 化学的进步(如碱基化学修饰和双脱氧测序的方法)实现的,而克隆和遗传学的革命则由分子生物学的重大发展(如聚合酶链式反应)引起。 RNA 的全基因组探测方法也在迅速发展。 在过去的 2-3 年中,已经开发出技术来探测体内整个基因组的 RNA 结构。 这是通过将使用过去 20-40 年开发的化学物质和酶修饰 RNA 的方法与过去 10 年开发的使用深度测序的方法相结合来实现的。 这些方法的持续改进至关重要,因为它将为基因组学和 RNA 生物学及其应用打开新的大门。
3.1 关于探测 RNA 结构的非全基因组方法的简短历史观点
简要考虑一下非全基因组方法来探测RNA结构的发展是有益的。当双链DNA的结构在1953年确定时,RNA似乎也可能发生碱基配对,尽管它是单链的。 1956 年,Rich & Davies (88) 解决了包含非生物聚合物 poly(rA) 和 poly(rU) 的双链 RNA 的纤维 X 射线衍射结构,该聚合物是具有类似于碱基配对和堆叠的右手双螺旋 双链DNA。 在 1960 年代,当研究人员发现单链 RNA 可以通过局部范德华 相互作用堆叠时,获得了对 RNA 结构的更多见解。 首先使用旋光色散 (13) 和核磁共振 (4) 的光谱方法描述了简单系统、同核糖聚合物和二核苷酸的 RNA 堆积。
transfer RNA(tRNA)的结构是第一个被确定为生物RNA的。 这种结构对 RNA 生物学和化学的影响怎么强调都不为过。 该结构是通过多种快速建立起来的方法确定的。 1965 年,tRNA-Ala 的序列被确定并手动折叠形成 A/U 和 G/C 碱基对(36),并推导出了三种可能的结构。 此后不久,其他 tRNA 序列被确定并折叠,在它们之间进行比较后,只发现了一个常见的折叠,即现在熟悉的三叶草。 这是 RNA 结构的第一个比较分析之一,这种比较,有时称为 “phylogenetic”,方法仍然是定义体内 RNA 碱基配对结构的有效方法 。
为了通过实验评估三叶草模型的二级结构,1972 年开发了寡核苷酸杂交方法,其中 tRNA 受到短四聚体的挑战。如果四聚体杂交,则 tRNA 的目标区域被认为是单链的 (113)。一年后,研究人员解开了 tRNA (47) 的晶体 X 射线衍射结构,发现它与这种二级结构一致。这种晶体结构为理解 RNA 结构和功能提供了范式转变。第一次可以看到二级和三级元素,如环、凸起和连接。这种结构表明,RNA 可以具有复杂的三维折叠,与蛋白质不同,从而对 tRNA 的功能和 RNA 的一般功能产生无数影响。例如,了解 tRNA 如何与氨基酸连接作为翻译的衔接子,预示着一种复杂的 RNA 分子如何识别核糖开关中的小分子并催化核酶中的反应。从 tRNA 的精液结构中揭示的 RNA 的分子复杂性预示了我们现在所知道的 RNA 的生物学复杂性。
这些早期研究中使用的方法fiber和晶体X射线衍射以及旋光色散和核磁共振光谱–不适用于以高通量方式研究RNA 或在体内。 RNA 的系统发育分析虽然功能强大且基于生物学比较,但需要来自遥远生命形式的基因组中广泛的、注释良好的序列信息。 因此,必须开发新的实验方法来直接探测 RNA 的结构。
3.2 Secondary Structure Probing
两大类试剂用于探测 RNA 结构:酶和化学品(表 1)。两者都提供了有关 RNA 结构各个方面的具体信息 (26)。用于探测 RNA 序列的 RNase 在定义的结构位置切割 RNA 序列。例如,RNase V1 在碱基配对区域切割 RNA,尽管结构特异性不是绝对的 (68)。相比之下,RNase S1 和 P1 通常会在非结构化区域切割 RNA,尽管这些酶可能会遗漏小突起或环 (114)。通过结合各种酶处理的结果,可以在很大程度上确定给定 RNA 的碱基配对和非结构化区域。然而,这种方法不适用于 RNA 结构的体内探测:这些大酶不是膜渗透性的,并且一些酶,例如 RNase V1,需要非生理 Mg2+ 浓度才能发挥活性 (49)。 因为 Mg2+ 也促进 RNA 折叠,如果试图将体外结构等同于体内结构,这可能会出现问题。
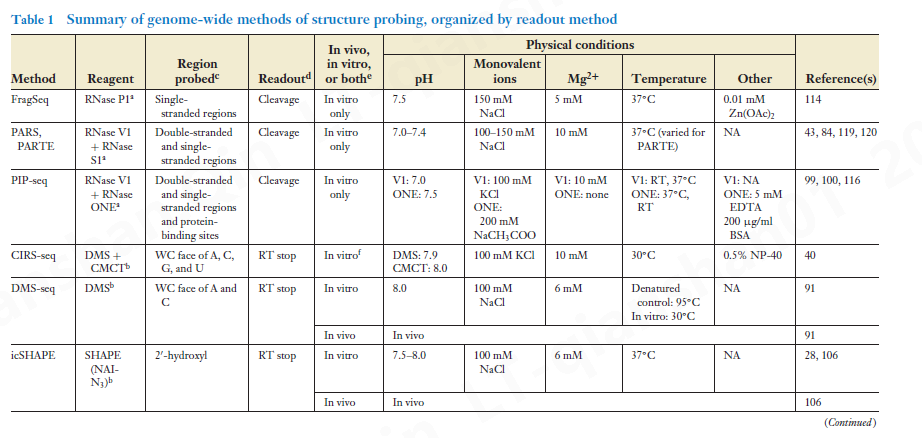
通常,核酸在化学上是不活泼的,这为遗传物质提供了稳健性。尽管如此,化学家已经设计出修饰碱基的 Watson-Crick (WC) 面、碱基顶部(Hoogsteen 面)或糖磷酸骨架上的官能团的试剂。在WC表面上,腺苷和胞苷与烷基化试剂如DMS(硫酸二甲酯)反应,而鸟苷和尿苷与碳二亚胺改性试剂CMCT [1-环己基-3-(2-吗啉代乙基)碳二亚胺甲基-对甲苯磺酸盐]反应。其他化学物质与糖-磷酸骨架发生反应,因此对单个核苷酸没有特异性。这些包括 SHAPE(通过引物延伸分析的选择性 2C-羟基酰化)试剂,可酰化糖的 2C-OH (125)。迄今为止,大多数研究只使用了其中一种化学试剂。然而,每种试剂都有其优点,使用不同试剂进行平行研究可能会给出最全面的 RNA 折叠视图。
一旦 RNA 被修饰或切割,无论是通过化学物质还是酶(表 1),都必须确定反应位点。不同的探测技术需要不同的读出方法。当 RNase 切割 RNA 骨架时,可以通过使用末端标记的 RNA 分析切割位点来确定 RNA 结构,通过 PAGE(聚丙烯酰胺凝胶电泳)或毛细管电泳分离,或使用引物在切割位点流出的逆转录;后一种方法适用于全基因组方法。 DMS 和 CMCT 等化学物质在核碱基的 WC 面上发生反应,通过阻止碱基配对来阻断逆转录。与糖-磷酸骨架反应的 SHAPE 试剂添加了也阻止逆转录的庞大基团。切割反应和逆转录 反应都可以直接转移到创建全基因组文库;因此,历史上基于凝胶的方法可以很容易地适应下一代测序方法,以高通量方式确定 RNA 的结构。
3.3 In Vitro Genome-Wide Methods
第一个全基因组 RNA 结构探测方法涉及酶探针,它只能在体外使用。 FragSeq(片段化测序)(114) 用 RNase P1 探测 RNA 结构,它切割单链区域(表 1)。 FragSeq 中报告的评分使用核酸酶的比率来控制处理以解释任何自然降解。
PARS(parallel analysis of RNA structure)使用 RNase V1 和核酸酶 S1 分别在双链和单链区域切割 RNA(表 1)。在二代测序之后,会生成一个 PARS 分数,它是 V1 与 S1 读数的比率,有效地报告了 RNA 的双链性 (43)。 PARS 的一个优点是它可以揭示在同一分子的不同拷贝中采用多于一倍的 RNA 区域,因为 V1 和 S1 都将在同一区域切割。
PARS 也适用于包括 PARTE(随温度升高并行分析 RNA 结构,(parallel analysis of RNA structure with temperature elevation),它允许通过测量不同温度下的 RNA 结构来计算 RNA 折叠能 (119)。这三种方法已使用生物 pH 值(pH 7-7.5),但在典型的体外结构探测浓度 Mg2+ (5-10 mM) 和一价盐 (100-150 mM NaCl) 远高于生理真核生物中 ~0.5-1 mM 的 Mg2+ 水平 (65) 和原核生物中 2 mM (71, 111)。
尽管许多有趣的发现来自对 RNA 结构的观察,但一个关键挑战是了解蛋白质在影响 RNA 结构中所起的作用。 PIP-seq(蛋白质相互作用谱测序,protein interaction profile sequencing)是为了部分解决这一挑战而开发的。 PIP-seq (99, 100) 使用 RNase V1 和单链 RNA 特异性 RNase (RNase ONE) 在两个 RNA 池上切割 RNA 的双链和单链区域,其中一个未经处理,一个未经处理 用蛋白酶 K 处理以去除所有蛋白质(表 1)。 尽管从概念上讲,这种方法提供了有关蛋白质结合和 RNA 结构的信息,但这种类型的分析可能存在固有问题,因为它假设 RNA 结构在去除蛋白质后保持静态。 RNA 结构也可能受到体外研究选择的溶液环境的影响。
RNA 结构也可能受到体外研究选择的溶液环境的影响。在 PIP-seq 中,用于单链和双链核酸酶探测的缓冲液不相同 (32)(表 1),这可能是有问题的。在一些研究中,游离 Mg2+,特别是促进 RNA 折叠的游离 Mg2+,在 RNase ONE 处理中不存在(通过添加 EDTA 去除),但在 V1 处理中不存在。因此,在 RNase ONE 探测与 V1 探测期间,给定的 RNA 物种可能具有不同的结构。总之,尽管 PIP-seq 可以提供有关蛋白质结合位点的信息,但在其用于推断 RNA 结构的应用中存在显着的注意事项。
基于化学的方法提供了一种在体外全基因组范围内探测 RNA 结构的替代方法。例如,CIRS-seq(RNA 结构的化学推断,随后是大规模并行测序,chemical inference of RNA structures followed by massive parallel sequencing)(40)使用 DMS 和 CMCT 分别与 A/C 和 G/U 的 WC 面反应。 CIRS-seq 因此提供了所有四个碱基的核苷酸分辨率。在 CIRS-seq 之前,全基因组分析仅使用 DMS,但这些应用是在体内(见下一节)。迄今为止,CIRS-seq 探测是在基于蛋白酶去除与 RNA 相关的蛋白质并在 pH 8.0 下使用 10 mM Mg2+ 并添加 0.5% NP-40 后进行的, 这可能会改变 RNA 的折叠(表 1)。
SHAPE 化学品修饰糖-磷酸骨架的糖,并提供所有四种核苷酸的柔韧性信息,其中柔韧性是碱基配对缺失的一般指标 (125)。这些化学物质已用于各种全基因组方法。通常,SHAPE 修饰会阻止逆转录。该方法的一种变体 SHAPE-MaP (98, 103) 使用突变分析来分析 SHAPE 反应在体外生物样条件下的位置,这些作者选择 pH 8.0、200 mM 一价盐和 3 mM Mg2+(表 1)。该方法修改了典型逆转录反应的溶液条件,以促进 SHAPE 修饰核苷酸的核苷酸突变(表 1)。然后通过下一代测序鉴定这些突变,其中突变位点被推断为原始折叠中的单链。这种方法的主要技术优势是聚合酶读取突变,从而允许在单个 RNA 分子中检测到多个突变。突变谱分析对于识别单个 RNA 中两个位置之间的相关性特别有用,但它的缺点是突变谱分析需要更深入的测序才能确定地称为单链核苷酸,因为在文库制备过程中可能包含与 SHAPE 加合物无关的突变。这种方法有可能成为一种体内探测方法,因为它结合了一种细胞可渗透的 SHAPE 试剂 (102, 105)。
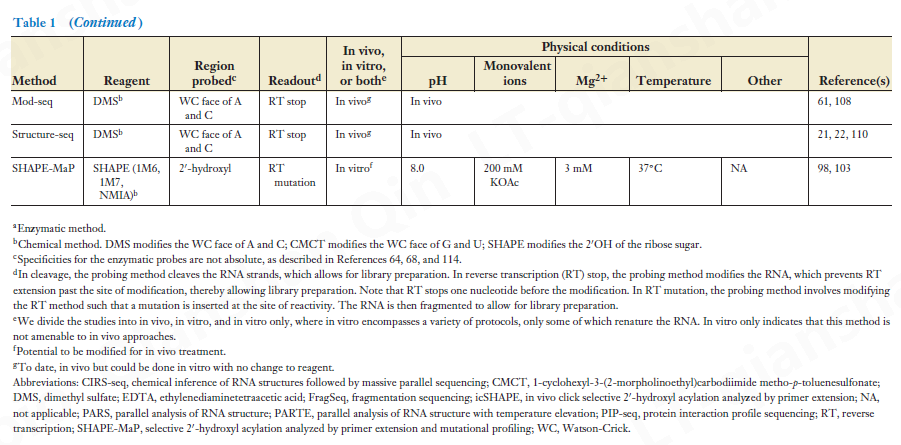
3.4 In Vivo Genome-Wide Methods
由于细胞环境对 RNA 结构的影响,普遍存在的结构在体外与体内通常不同 (129)。因此,尽管可以从体外全基因组 RNA 结构分析中获得一些信息,但在体内 RNA 结构探测中回答生物学问题很重要。目前,唯一能与核苷酸的 WC 面反应并已用于体内全基因组方法的化学物质是 DMS。尽管 DMS 仅与 A 和 C 发生反应,但这种反应性与 SHAPE 反应性在 RNA 结构预测中的表现一样好 (17)。几乎同时开发的 Structure-seq (22)、DMS-seq (91) 和 Mod-seq (108) 使用 DMS 在体内与 RNA 反应。 RNA 提取后,这些方法使用逆转录来识别由扩展中的终止标识的反应位点(表 1)(对于这三种方法之间差异的更全面分析,请参见参考文献 54)。在这些方法中,Structure-seq 具有一定的优势:它在实验流程的早期将 RNA 转换为 DNA,因此使用更不稳定的核糖核酸需要更少的操作,并且具有强大且用户友好的计算流程 (110)在 Galaxy 环境 (https://usegalaxy.org/) 中提供访问权限。
如上所述,某些 SHAPE 化学品也可用于绘制体内 RNA 结构图 (105)。一种使用 icSHAPE(体内点击 SHAPE)反应 (106) 的方法通过生物素-链霉亲和素分离富集了 SHAPE 修饰的 RNA 片段(表 1)。这种分离是可能的,因为无铜点击(copper-free click)反应会产生一种修饰的 SHAPE 化学物质,附着在生物素化的部分上。由于 SHAPE 方法探测糖磷酸骨架,因此 icSHAPE 提供了有关所有四种核苷酸的灵活性的信息。
在体内研究 RNA 结构很重要,但目前可用的方法存在一些限制。特别是,没有已知的化学物质可以修饰双链区域。因此,直接信息仅适用于基于化学反应性研究的单链区域,包括体外和体内。蛋白质结合提供的对反应性的保护进一步加剧了这个问题。因此,化学探测研究中缺乏反应性可能是由于 RNA 部分是双链的,也可能是由于蛋白质结合该区域。然而,反应性的存在是单链性的高度可靠指标 (22),仅此信息就可以极大地抑制 RNA 结构的预测。因此,尽管全基因组方法学中的某些领域需要进一步发展(在下面的挑战和未来方向中讨论),但当前的技术非常有价值,因为它们可以获取有关系统的大量知识;例如,单链区域的反应性变化提供了有关 RNA 转换以及蛋白质结合和解除结合的信息。此外,通过比较跨环境条件或发育阶段的 DMS 反应性读数,可以了解更多关于 RNA 结构和蛋白质结合变化的信息。
3.5 Methods to Identify Natural RNA Modifications Genome-Wide
尽管 tRNA 和核糖体 RNA (rRNA) 中存在的天然修饰已为人所知多年 (34),但仅自 2014 年以来,才描述了全基因组修饰。最近,五种RNA修饰: N6-甲基腺苷(m6A)(23、62)、假尿苷(M-OM-H)(14、93)、N1-甲基腺苷(m1A)(24、59)、5 -甲基胞嘧啶 (44, 45, 107) 和 2'-O-核糖甲基化 , 在全基因组范围内鉴定;前三个也与mRNA结构方面有关。由于这些修饰是共价变化,因此它们是稳定的;因此,体外研究可以准确地报告它们的位置。m6A 不修饰 WC face,最初是使用基于抗体的技术 (23) 来拉下带有修饰的 RNA 区域,而 ψ 是由与修饰 CMC(N-环己基-N9-(2-吗啉代乙基)-碳二亚胺甲基-对甲苯磺酸盐)的化学反应提供逆转录终止 (14, 93)。 m1A 已通过液相色谱 质谱法在 mRNA 中被完全鉴定,而基于抗体的 pulldown 结合下一代测序已被用于在全基因组范围内绘制包含 m1A 的序列 (24, 59)。 m1A修改WC面;因此,它也可以通过在逆转录中诱导终止和错配组合来识别 (112)。下文描述了与 ψ、m6A 和 m1A 相关的结构特征的生物学意义。
四、INSIGHTS FROM EXPERIMENTAL GENOME-WIDE STUDIES OF THE RNA STRUCTUROME
使用来自转录组分析的数据,正在重新评估和扩展关于 RNA 结构-功能关系的单基因研究的结果,特别是对于 mRNAs。这些研接下来,我们总结了 RNA 结构组的元特性,这些元特性在全基因组研究中有明确的统计支持,但其功能影响和分子基础通常仍然知之甚少。随后的部分总结了与 RNA 修饰、加工、稳定性和翻译相关的元属性。
该领域处于起步阶段。迄今为止,仅为少数几个系统生成了实验结构组。第一个要描述的 RNA 结构组是 HIV-1 RNA 基因组的结构组,Weeks 及其同事使用他们的 SHAPE 方法在体外对其进行了探测(125)。自 2009 年发表开创性文章以来,已经在酵母、大肠杆菌、拟南芥、果蝇、秀丽隐杆线虫以及小鼠和人体组织和细胞系中对 RNA 结构进行了全基因组检测(表 2)。随着对更多物种、系统和条件的分析,重要的是要确定哪些结果强化了以下部分中描述并总结在表 2 中的原则,哪些结果可能揭示迄今为止未设想的 RNA 结构控制机制。图 3 提供了与 RNA 结构相关并通过全基因组研究揭示的现象的概述。
4.1 RNA 结构组的元特性 Meta-Properties of RNA Structuromes
完整的 RNA 结构组包括所有类型 RNA 的结构数据,包括编码和非编码。一些全基因组研究比较了作为非编码 RNA 与 mRNA 特征的结构程度。通过使用 PARTE 分析 RNA 在体外展开的温度依赖性,Chang 及其同事 (119) 发现酵母非编码 RNA(rRNA、tRNA、小核仁 RNA、小核 RNA)在比 mRNA 更高的温度下熔化。 mRNA 的 Tm 差异最大,与其高度多样化的性质一致。与 mRNA 相比,在拟南芥体外结构组的 rRNA、tRNA、microRNA、小核仁 RNA 和小核 RNA 中发现了更多的双链,表明二级结构更大(130)。鉴于它们的生物学作用和作用机制,预计非编码 RNA 中的稳健结构。
包括 poly(A) 选择步骤的全基因组结构探测方法导致小 RNA 的代表性不足,并且大多数全基因组结构分析都集中在 mRNA 上。 mRNA 结构组的一个强大的元属性是预测结构和/或反应性的三重重复模式,其存在于 mRNA 的编码序列 (CDS) 上,但在 UTR 中不存在。这种现象在计算机结构组 (95)、体外 (19, 40, 43, 119) 和体内 (22, 106) 中反复出现,并且已在多种生物体中观察到,包括大肠杆菌 (19)、酵母 ( 43, 119)、拟南芥 (22) 和小鼠 (40, 106) 和人 (120) 细胞系。三重重复(triplet repeat)完全是序列固有的基础,由影响RNA折叠和反应性的实验或细胞条件强加,和/或以某种方式从文库构建中的偏差中出现尚未有待解决,也不知道为什么自然选择可能偏爱这种模式。我们假设在功能上,三重重复序列可以提供一个寄存器,最大限度地减少翻译过程中核糖体的滑动(即,核糖体推进两个或四个核苷酸而不是三个),但是目前这只是猜测。三重重复不是简单地由核糖体结合强加的,因为它可以在脱蛋白的体外样品中看到(表 2)。 铁汉 15:43:12 在多个体内 mRNA 结构组中观察到但具有未知功能后果的另一个元特性是 CDS 和 UTR 之间平均反应性/RNA 结构的总体差异。CDS 和 UTR 之间的相对结构范围似乎以生物体特异性方式变化(见表2)。此外,这种关系可能在不同的细胞区室中有所不同。例如,在拟南芥中,总 mRNA 结构的体外 (57) 和体内 (22) 分析均显示 CDS 比 UTR 更具结构化。相比之下,核定位转录本的体外分析 (32),可能富含前 mRNA,显示 CDS 的结构不如 UTR。从表面上看,这些结果表明,与前 mRNA 相比,RNA 加工事件增加了加工 CDS 中的平均结构或减少了 UTR 中的平均结构。为什么进化可能选择不同生物体或细胞区室中的不同结构模式仍然是一个有趣的问题。

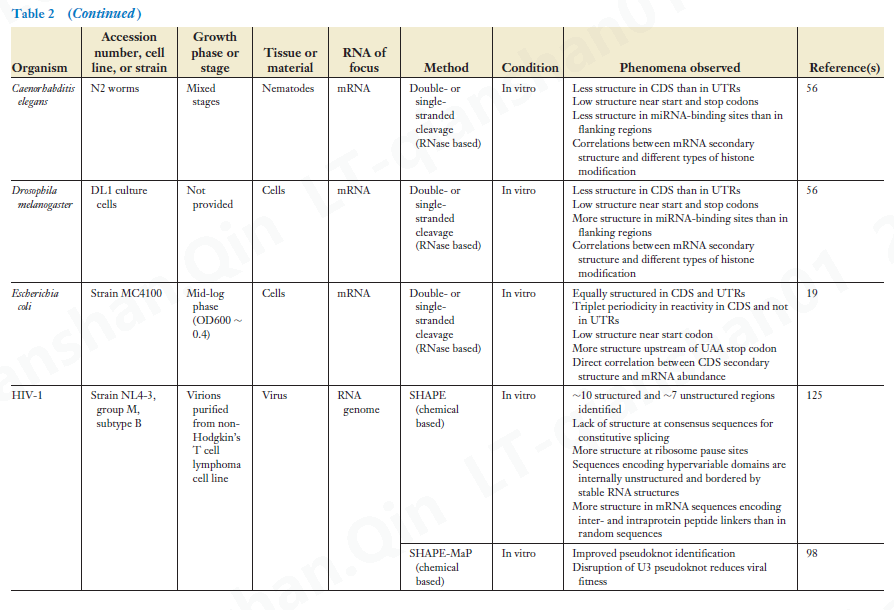
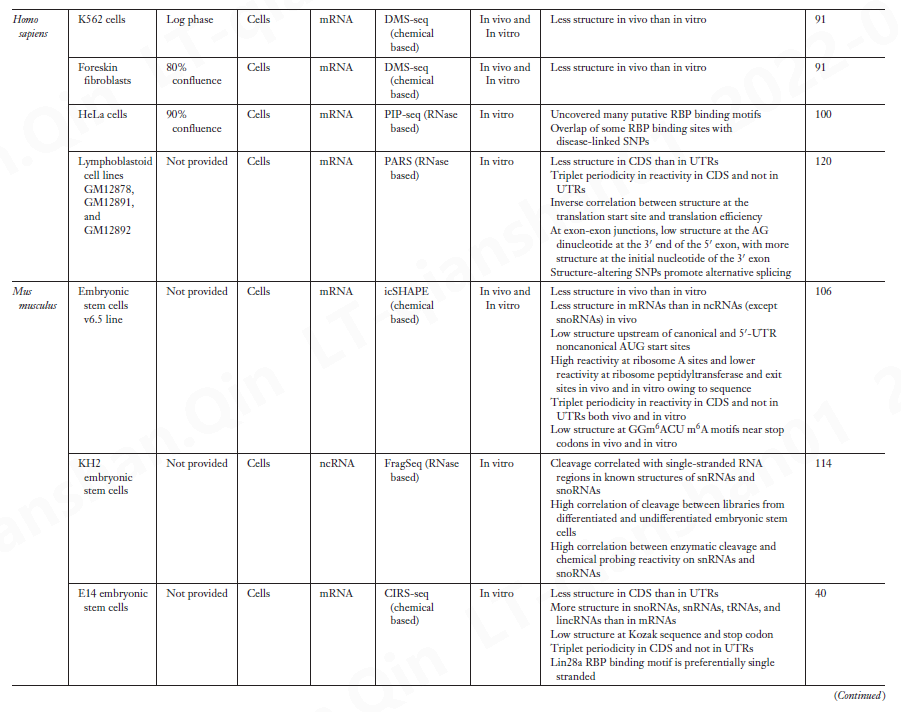
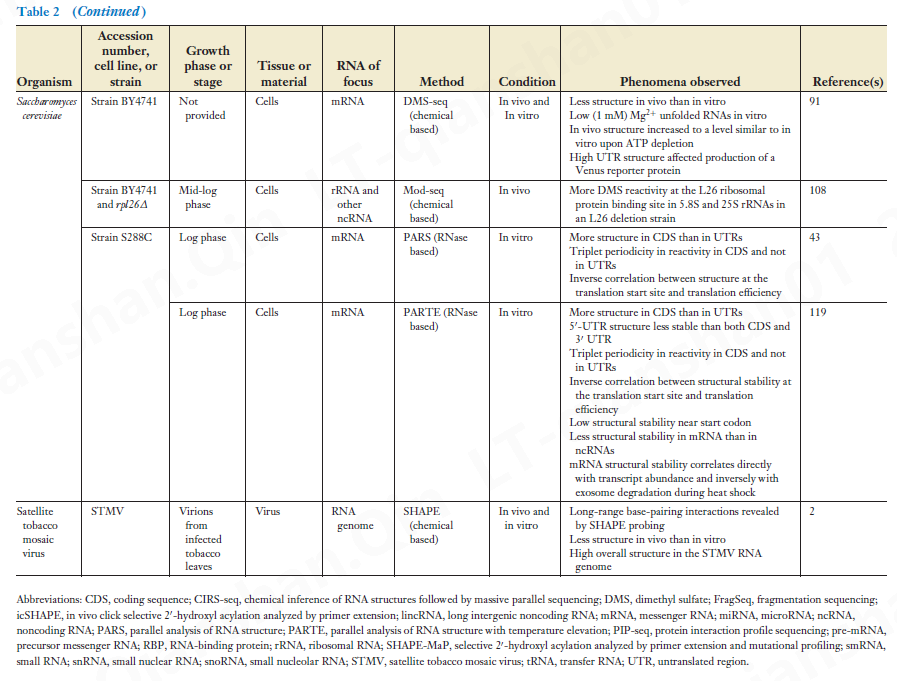

另一种类型的荟萃分析涉及体内与体外结构组的比较。 Weissman 实验室的荟萃分析表明,在从体外结构组研究中得出结论时应谨慎行事 (91)。他们对酵母的体外和体内结构组进行了直接比较,发现体内结构组的整体结构不如体外结构组 (91)。虽然这个结果最初是出乎意料的,但回想起来可能并不那么令人惊讶,因为大多数体外分析都以高 Mg2+ 浓度为代表,这往往会促进折叠。事实上,Rouskin 等人。 (91) 发现当 Mg2+ 的缓冲液浓度降至 1 mM 时,体外结构组发生显着展开。他们进一步证明,体内实验性的 ATP 消耗导致了更多的体外 结构组。该结果表明,能量依赖性过程,例如 RNA 的解旋酶解旋,在减少体内整体 RNA 结构中起重要作用。斯皮塔莱等人。 (106) 随后证明,小鼠胚胎干细胞系在体内的 RNA 结构也比在体外显示的少,特别是在 mRNA 中。当在病毒体内或体外进行探测时,卫星烟草花叶病毒的 RNA 基因组也观察到了类似的现象 (2)。
4.2 与 RNA 结构和 mRNA 加工相关的元特性
新生 mRNA 需要经过许多加工步骤,包括加帽、剪接和多聚腺苷酸化。在从细胞核输出期间和之后,还会发生更多水平的调节,最突出的是与亚细胞定位、翻译和周转相关的调节。与蛋白质的结合 (30, 41) 可能是短暂的,或者就像核糖体一样,寿命更长。正如先前的单基因研究和越来越多的 RNA 结构组分析所揭示的那样,RNA 结构影响 RNA 加工的所有阶段。相反,在单个 RNA 分子的整个生命周期中,亚细胞环境和蛋白质结合的变化会以高通量 RNA 结构研究刚刚开始揭示的方式影响 RNA 结构。
替代多聚腺苷酸化(Alternative polyadenylation)。
替代多聚腺苷酸化通过在 3UTR 内的替代位点或很少发生在内含子或外显子内的替代位点发生切割和多聚腺苷酸化。由此产生的序列差异会影响 mRNA 加工和利用的所有方面,包括剪接、亚细胞定位、翻译和降解 (27, 77)。替代多聚腺苷酸化在哺乳动物和植物中都很常见。尽管已经确定了替代多聚腺苷酸化位点的序列特征 (27),并且已经提出二级结构会影响多聚腺苷酸化效率 (48) 和位点选择 (33),但仅在两个实验性全基因组结构中评估了 RNA 结构的可能作用迄今为止的研究 (22, 32)。在植物中,50- 80% 的前 mRNA 可以进行选择性多聚腺苷酸化 (39, 97)。使用拟南芥幼苗的体内结构序列分析,Ding 等人。 (22) 发现已知进行选择性多聚腺苷酸化的位点在核苷酸 -22 至 -15(即切割位点上游)处表现出 DMS 反应性低的区域和 DMS 反应性高的区域,指示低结构,在核苷酸 1 至 5(即,切割位点的交叉和下游)。他们还发现,这种模式不仅仅是由于核苷酸组成。然而,核拟南芥结构组的体外分析未能揭示与替代多聚腺苷酸化位点相关的结构特征
。。。。
五、总结
总结要点:
- RNA 折叠成复杂的二级和三级结构。
- 活细胞中的 RNA 折叠受多种环境和物理因素的影响。 RNA 和 RNA-蛋白质相互作用的修饰也会影响 RNA 结构。
- 可以使用基于核酸酶和化学探测的体外方法来研究全基因组 RNA 二级结构。
- 最近开发了用于 RNA 二级结构的全基因组化学探测的体内方法。
- 存在探测核碱基和糖-磷酸骨架的方法。
- 全基因组结构探测方法正在揭示与 mRNA 加工各个方面相关的 RNA 结构的新元特性,包括多聚腺苷酸化、剪接、共价修饰、翻译和转换。
- RNA结构的变化构成了基因调控的一个基本方面,以响应我们刚刚开始理解的不断变化的环境条件。
未来问题:
- 体外 RNA 结构组的哪些特征忠实地反映了体内结构组,哪些不?
- 类、组织和发育阶段的M-bM-@M-S特异性体内RNA结构组将揭示哪些新信息?
- RNA 结构组的可塑性如何帮助生物体应对生物和非生物环境压力?
- 完整的 RNA 表观转录组如何影响 RNA 的结构和功能?
- 我们如何识别额外的 RNA 修饰及其对全基因组 RNA 结构的影响?
- 我们如何进一步改进 RNA 的体内结构探测?我们如何才能更好地区分体内和全基因组的 RNA 碱基配对和蛋白质保护?
- 我们能否开发出在体内探测 RNA 分子中所有关键原子的方法?
- 我们如何才能最好地预测活细胞中给定 RNA 的结构集合?
- 我们如何才能最好地设计针对传染病的基于 RNA 的疗法?
- 我们如何才能最好地设计药物来修复导致遗传病的 RNA 结构异常?
參考資料
- Home Annual Review of Genetics Volume 50, 2016 Bevilacqua, pp 235-266
Save Email Share
Genome-Wide Analysis of RNA Secondary Structure 。 https://www.annualreviews.org/doi/full/10.1146/annurev-genet-120215-035034#article-denial
