【6.3】SPROUT
对Cas9诱导的DNA切割后修复结果的了解仍然有限,尤其是在原代人类细胞中。 我们在人类原代T细胞中1,656个目标基因组位点上对修复结果进行测序,并使用这些数据来训练机器学习模型,我们将其称为CRISPR修复结果(SPROUT)。 SPROUT可以准确预测核苷酸插入和缺失的长度、概率和序列,并将有助于在具有治疗意义的原代人细胞中设计SpCas9指导RNA。
原代T细胞是用于治疗性基因组编辑的有前途的细胞类型,因为它们可以离体有效地工程改造并过继转移给患者。但是,缺少有关人类原代细胞中Cas9依赖性编辑的基因组结果的详细信息。在这里,我们系统地表征了化脓性链球菌(Streptococcus pyogenes)Cas9(SpCas9)在来自18位健康献血者的原代T细胞中的修复结果(补充图1)。
靶向测序已应用于CD4 + T细胞中559个基因中的1,656个独特基因组位置。指导RNA(gRNA)与SpCas9结合以组装核糖核蛋白复合物(RNP),并电穿孔进入T细胞。恢复和扩增6天后,从细胞中分离出DNA,并对每个位点周围的180至260个碱基对区域进行PCR扩增和测序(图1)。我们使用CrispRVariants4(图1)从生成的扩增子文库中量化了每个靶位点修复结果的分布。共有31%的reads包含以剪切位点为中心的小片段,平均缺失长度为13个碱基对。我们还发现20%的reads在剪切位点有插入,而这些插入中的95%恰好是1个核苷酸(补充图2)。只有0.008%的read同时包含插入和删除。
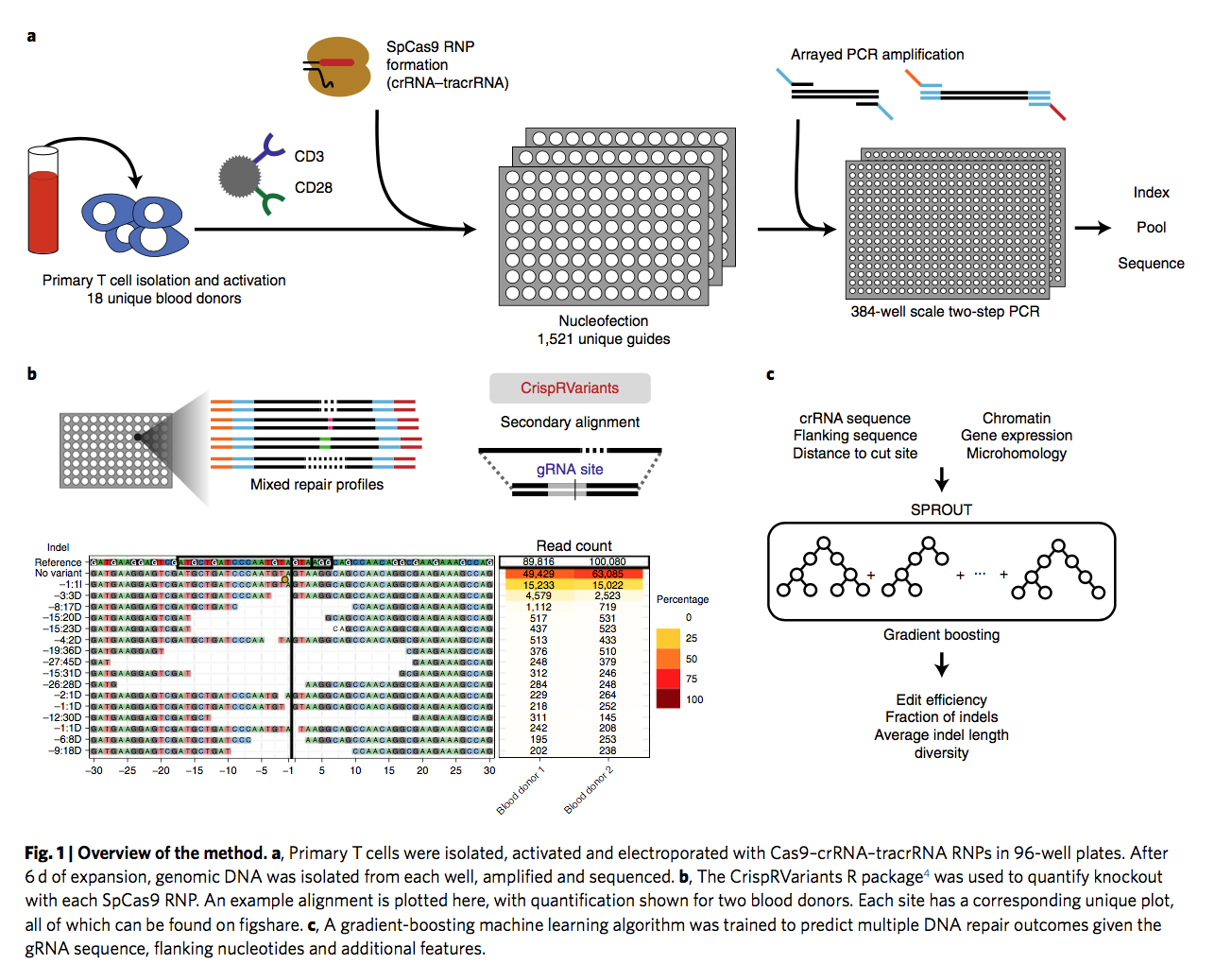
每个目标位点平均有98个离散的修复结果,其频率大于1,000次读取中的1个,并且不同位点的插入和缺失的比例和长度分布差异很大。每个目标部位的修复结果在供体之间相似,但不同目标部位之间的差异很大(图2a)。所有site之间修复结果的比较表明,来自单个目标站点的重复编辑实验的结果彼此之间的相似性远高于来自不同站点的结果(图2b和补充图3)。

我们假设切割部位之间修复结果的差异主要是由于切割部位附近的序列差异引起的。为了对此进行测试,我们开发了一种机器学习模型SPROUT来预测SpCas9修复结果(图1)。该模型将间隔序列的20个核苷酸加原间隔子相邻基序(PAM)作为输入,并使用梯度增强(gradient boosting )在该核苷酸上训练决策树的整体。在每个目标位点,该模型预测插入/缺失(图2c)或缺失(插入的1-fraction),以及插入和缺失的平均长度(图2d)的indel突变读段。我们包括了插入缺失突变体reads和总reads的比例,以分开对编辑效率的依赖性。在原代T细胞中独立的304个靶位点集上,SPROUT能够准确预测 fraction of indel mutant reads with an insertion (R2 = 0.59 and Spearman rank = 0.81) and the fraction of total reads with an insertion (R2 = 0.40 and Spearman rank = 0.68; Fig. 2c and Supplementary Figs. 4 and 5). SPROUT还能够预测目标是否具有,高(大于60%),中(40-60%)或低(小于40%)分数(图2d)的移码修复结果,其准确度为0.6。
SPROUT也可以用于计算机gRNA设计。对于具有多个gRNA的532个基因中的每个基因,我们使用SPROUT的预测将基因中的靶标从最可能发生移码修复结果的可能性降到了最低可能性。 SPROUT正确地鉴定了54%的基因中表现最佳的移码gRNA,并正确预测了38%的基因的完整排名(补充图6)。我们进一步调查了SPROUT是否可以正确选择基因中哪个SpCas9目标位点最有可能在插入序列上比插入序列富集。对于每个基因,我们使用来自SPROUT的预测对目标位点按插入的indel突变体读段的预测分数进行排序。对于73%的基因,SPROUT正确选择了最短的短gRNA,而对于60%的基因,它正确地预测了所有候选gRNA的插入比例的完全排名,这与随机猜测带来的显着改善相对应(补充图6, P <10的-10次方)。
预测信号主要位于切割位点左右两侧的3个核苷酸上。 -1位置(在切割位点的5’附近)影响最大(图2e和补充图7)。这与以前的观察结果一致,即该核苷酸在许多切割位点重复,这被认为是Cas9产生的单碱基突出端修复的结果(参考文献7)。在该位置G或C核苷酸的存在降低了插入比例,分别有7%和10%的插入缺失突变体reads。相比之下,在该位置存在A或T核苷酸的比例分别增加到23%和26%。在确定插入或删除的结果比例中,+ 3位置也很重要(补充图7)。与C和T分别为16%和15%的比例相比,此位置的A和G核苷酸分别将插入比例增加到25%和23%。均聚物(homopolymers)(两个或多个相同核苷酸的序列)在切点附近,增加了缺失的比例(P <0.02)。例如,在切割位点附近有G均聚物的靶标中有92%的插入缺失突变体缺失,而当切割位点处没有均聚物时,77%的reads具有缺失(补充图8),microhomology-mediated end joining的反映。
接下来,我们通过使用在T细胞数据上训练的SPROUT模型来预测该算法在其他人类细胞类型中的SpCas9修复结果,从而评估了算法对序列和细胞类型特定特征的鲁棒性。 我们重新分析了在HEK293,K562和HCT116细胞中测试的96个独特靶位点的公开靶向测序数据。 这96个目标不同于用于训练SPROUT的1,521个站点,因此构成了新的测试数据。 当预测带有插入的插入缺失突变体的分数时,SPROUT的准确度为R2 = 0.40,而预测带有插入的预测缺失总数的R2 = 0.23。 跨不同细胞类型的SPROUT相对较高的性能进一步表明,在分裂细胞内SpCas9裂解后,影响修复结果的主要因素是切割位点附近的核苷酸序列。
我们系统地比较了SPROUT与最近开发的两种预测SpCas9修复结果的方法inDelphi和FORECasT。在三种算法上进行了比较:
- the fraction of repair outcomes with a frameshift,
- the repair precision (defined as 1−indel diversity)
- the fraction of indel reads that are insertions
为了严格比较算法,我们在训练完所有三个模型后生成了三个新的SpCas9修复数据集。我们收集了两个新的原发性T细胞SpCas9修复结果数据集:首先是针对CXCR4基因的32个位点,然后是来自91个免疫相关基因的182个唯一位点。每个位点在多个供体之间复制。这些位点不同于用于训练SPROUT的T细胞位点,因此可作为独立验证。在这些方面,在维修预测任务中,SPROUT的性能明显优于InDelphi和FORECasT(补充图9; P <0.01)。我们还从编辑的人诱导多能干细胞(iPSC)的相同CXCR4站点收集了修复数据。没有在这些站点上对SPROUT进行培训,也没有看到来自人类iPSC的数据,因此这对另一种与治疗相关的细胞类型构成了强有力的考验。同样,对于三个预测任务中的每一个,在人类iPSC的数据上,SPROUT比inDelphi和FORECasT更准确(P <0.05;补充图9)。这些结果表明,SPROUT是预测T细胞和人iPSC中SpCas9编辑结果的最先进方法,这两种细胞正在共同努力利用CRISPR进行工程细胞疗法。
在90%的T细胞SpCas9目标位置中,我们在修复结果序列数据中发现了长的(> 25个碱基对)DNA插入。跨位点,有40%的长插入片段与人类基因组对齐,0.07%的为indel-containing reads。在对齐的长插入片段中,有36%与SpCas9目标位点对齐在同一条染色体上,其中27%与目标位点的1kb以内对齐(补充图10)。其余插入物与不同染色体上的位置对齐,这些位置被富集,可与目标位点进行Hi-C相互作用(P <10的-5次方;补充图11-13)。这些发现提示了一种可能的模型,通过该模型可以在DNA修复过程中插入与切割位点物理邻近的基因组区域。最近的报告表明,细胞可能会因SpCas9的裂解而发生基因组重排[11,12],尽管鉴于所用的细胞类型和其他变量,应谨慎解释这些发现。 CRISPR在原代T细胞和其他人类细胞中的潜在治疗应用,值得进一步研究基因组编辑过程中插入和其他重排的机制和普遍性。
二、方法
试验方法略
T细胞和iPSC数据摘要。
这项研究涉及来自18个个体的T细胞的3989个DNA修复图谱。这些结果靶向人类基因组中549个基因中的1,521个独特位点。选择了靶向编码HIV相互作用蛋白的基因的gRNA。为每个基因选择了Dharmacon Edit-R预先设计的敲除文库中的前三个gRNA。从每个平板上都选择了从Edit-R库中选择的三个不同的非靶向对照,以及三个经过验证的,定制设计的gRNA,这些gRNA已知可以高效敲除CXCR4,CDK9和LEDGF基因。 RNP敲除平均在来自不同献血者的独特原代T细胞中重复两次(补充图1)。修复结果在整个献血者的重复过程中进行平均,来自每个靶位点的DNA修复结果数据已存储在figshare( https://figshare.com/projects/Systematic_characterization_of_genome_editing_in_primary_T_cells_reveals_proximal_genomic_insertions_and_enables_machine_learning_prediction_of_CRISPR-Cas9_DNA_repair_outcomes/37166 )
通过设计沿CXCR4基因平铺的gRNA,生成了两个附加的验证数据集,这些gRNA在新的原代T细胞和iPSC供体中一式三份地重复进行。过滤质量后,使用与原始T细胞数据相同的过程,分析了修复结果测序数据中原代T细胞中的32种新gRNA和人iPSC中30种新的gRNA。使用靶向182个与91个免疫相关基因的起始密码子接近的基因座的gRNA,在原代T细胞上生成了第三个附加验证数据集。每个gRNA在六个独特的供体上进行了测试。这些新的验证实验中的所有目标站点都不同于用于训练SPROUT的sites。
HCT116,HEK293和K562数据摘要。
根据与T细胞数据相同的程序分析了来自其他三种细胞类型(HEK293,K562和HCT116; BioProject PRJNA326019)的已发布测序数据 用于验证机器学习模型。 我们从手稿中使用的数据集包括48小时后来自人类基因组96个独特切割位点的RNP基因敲除。
统计分析
我们使用梯度提升(gradient-boosting)算法来训练SPROUT。 梯度提升是一种聚合模型,可迭代地学习基础分类器的加权集合。 SPROUT使用决策树作为基础分类器。 树的深度和数量是算法的超参数,我们通过交叉验证来设置。 在SPROUT中,通常使用20–200个决策树,每个决策树的深度为3–20层,具体取决于预测任务。 补充图14中列出了评估为包含在SPROUT中的所有功能的完整列表。
我们使用了五重交叉验证来训练SPROUT。我们将T细胞中的独特切割位点(总共1,521个)随机分成五折并训练了SPROUT 五折中的四折。然后,我们在剩下的看不见的五折(304个剪切位)上测试了SPROUT的性能。我们重复了十次随机数据分割过程,并报告了十次随机重复的预测性能的平均值和标准差。我们使用不同大小的训练集进行了训练,并且SPROUT的性能出现了 使我们当前在T单元上的数据大小饱和(补充图15)。我们使用确定系数(R2)评估了回归任务的预测性能(即,prediction of the fraction of total or indel mutant reads with insertion or deletion and the editing efficiency)。我们还使用分类器的准确性评估了分类任务的预测性能(即,预测平均插入或缺失长度或多样性,是否大于或小于分布的中位数)。修复多样性定义为读取中修复结果分布的熵。高度的多样性表明该site的修复结果更具可变性。naive(或随机的)猜测在预测正确的输出标签时将准确度达到50%。
对于在其他三种细胞类型(HCT116,HEK293和K562)上评估的模型,我们在完整的T细胞数据(1,521个切割位点)上训练了SPROUT,并在其他细胞类型上测试了该模型的性能。 我们没有使用其他细胞类型特有的功能来微调SPROUT来量化模型的健壮性。 对于分类任务,我们使用cell type分布的中位数来设置阈值。 其他详细信息包含在补充说明中。
与InDelphi和FORECasT的比较
我们在以下四个基准数据集上将SPROUT与InDelphi和FORECasT进行了比较:
- 在SPROUT训练期间未使用的保持性T细胞测试数据集;
- 横跨CXCR4的32个SpCas9目标位点的primary T细胞数据集;
- 91个免疫相关基因中182个SpCas9靶位点的主要T细胞数据集;
- 正文中详述的来自人类iPSC的新数据集。
数据集2、3和4是在开发SPROUT之后收集的,它们由新的基因组基因座和新的供体组成,而这在SPROUT培训期间是看不到的。
我们使用了 http://indelphi.giffordlab.mit.edu/ 提供的训练有素的inDelphi模型,以在基准数据集上测试此方法的性能。 将U2OS设置为输入细胞类型(在提供的细胞类型中,最接近T细胞的结果)。移码和精确度是直接从网站图形界面下载的。我们使用inDelphi论文中提出的修复精度定义,该定义是减去缺失长度频率分布的熵。我们为每个实验下载了修复结果,并使用脚本查找了带有插入片段的片段。
我们使用网站https://partslab.sanger.ac.uk/FORECasT 来评估FORECasT方法中的移码。测量精度和分数 带有插入片段的reads数,我们使用了提供的经过训练的模型的批处理模式 https://github.com/felicityallen/SelfTarget 和后处理脚本。对于移码和精度,我们将预测值设定为阈值,并将其分为“高”和“低”类别,并报告该方法正确预测的类别百分比。对于插入的分数,我们报告R2值。
从SPROUT中提取的核苷酸特征解释
为了测量梯度增强模型中各个特征的重要性,使用了信息增益概念。 与特征相关的信息增益可衡量在根据特定特征拆分数据集后熵的降低。 较高的信息增益对应于更具预测性的功能。 我们还根据在数据上训练的线性回归模型的系数的符号确定了每个特征(富集或耗竭)的影响。 注意,该算法完全看不到切割部位的实际位置。 另外,核苷酸的特征重要性(例如G)显示出交替的模式。 我们推测,以交替模式( alternating patterns)丰富插入结果的原因之一是均聚物(homopolymer)效应。 据观察,均聚物(一个碱基的重复产生相同核苷酸的长序列)有利于缺失结果
根据所需的修复结果对gRNA进行排名。
我们根据所需修复结果的产生情况评估了SPROUT对gRNA进行排名的能力。使用两个输出来训练回归,即indel read的分数和总reads的分数。在训练了400个基因后,该模型用于预测针对149个不同基因的一组gRNA的插入和缺失的比例。我们仅评估了数据集中具有多个gRNA的基因(总共149个基因的142个测试基因)上的gRNA的排名性能。然后根据插入和缺失的分数对每个基因中的gRNA进行排名,并评估观察结果与预测排名之间的排名相关性。
使用Kendall的τ排名系数和完全正确的预测百分比来衡量效果。肯德尔的τ排名系数用于衡量观察结果与预测排名之间的差异。肯德尔的τ系数是介于-1和1之间的排名量度,其中1表示排名完全匹配,0表示不存在排名相关性,-1表示排名相关性完全相反。补充图6总结了来自T细胞的保留基因中的gRNA和来自其他三种验证细胞类型(HCT116,HEK293和K562)的gRNA的排名结果。
从修复结果(repair outcomes)中提取并对齐长插入数据。
为了获得插入数据,解析所有1,521个剪切位点的修复结果,并选择长度至少为25个碱基对的插入序列进行读取,从而产生总共22,495个以插入位点为中心的独特插入。 在默认条件和输入参数下,使用BLAST算法(blastn命令; https://blast.ncbi.nlm.nih.gov/Blast.cgi/ )将所有插入片段与人类基因组对齐。 对于具有多个对齐方式的情况, 选择了比对得分最高的site。 共有8,946个与人类基因组对齐的独特插入。
报告摘要。
有关研究设计的更多信息,请参见与本文链接的《自然研究报告摘要》。
Data availability
All the raw data and analyses are openly available through SRA (BioProject PRJNA486372) and figshare ( https://figshare.com/projects/Systematic_characterization_of_genome_editing_in_primary_T_cells_reveals_proximal_genomic_insertions_and_enables_machine_learning_prediction_of_CRISPR-Cas9_DNA_repair_outcomes/37166 ), respectively.
Code availability
https://zou-group.github.io/SPROUT
参考资料
- NATuRE BIoTECHNoLoGy | VOL 37 | SEPTEMBER 2019 | 1034–1037 | www.nature.com/naturebiotechnology. Large dataset enables prediction of repair after CRISPR–Cas9 editing in primary T cells
